Synergistic effects of methyl 2-cyano-3,11-dioxo-18beta-olean-1,-12-dien-30-oate and erlotinib on erlotinib-resistant non-small cell lung cancer cells
2022-01-19EonyNottinghmElizethMzzioSunilKumrSurpneniShlluKutlehriArindmMondlRmeshBdisStephenSfeArunRishiMndipSingh
Eony Nottinghm,Elizeth Mzzio,Sunil Kumr Surpneni,Shllu Kutlehri,Arindm Mondl,Rmesh Bdis,Stephen Sfe,Arun K.Rishi,Mndip Singh,*
aDepartment of Pharmaceutics,College of Pharmacy and Pharmaceutical Sciences,Florida A&M University,Tallahassee,FL,32307,USA
bDepartment of Veterinary Physiology and Pharmacology,College of Veterinary Medicine,Texas A&M University,College Station,TX,77843,USA
cJohn D.Dingell VA medical Center and Department of Oncology,Wayne State University,Detroit,MI,48201,USA
Keywords:
Transcriptomic analysis
Combination therapy
Drug resistance
Erlotinib
Epidermal growth factor receptor
Peer review under responsibility of Xi’an Jiaotong University.
A B S T R A C T
Non-small cell lung cancer(NSCLC)is often characterized by an underlying mutation in the epidermal growth factor receptor(EGFR),contributing to aggressive metastatic disease.Methyl 2-cyano-3,11-dioxo-18beta-olean-1,12-dien-30-oate(CDODA-Me),a glycyrrhetinic acid derivative,reportedly improves the therapeutic response to erlotinib(ERL),an EGFR tyrosine kinase inhibitor.In the present study,we performed a series of studies to demonstrate the efficacy of CDODA-Me(2μM)in sensitizing HCC827R(ERL-resistant)cells to ERL.Herein,we first established the selectivity of ERL-induced drug resistance in the HCC827R cells,which was sensitized when ERL was combined with CDODA-Me(2μM),shifting the IC50from 23.48 μM to 5.46 μM.Subsequently,whole transcriptomic microarray expression data demonstrated that the combination of ERL+CDODA-Me elicited 210 downregulated genes(0.44% of the whole transcriptome(WT))and 174 upregulated genes(0.36% of the WT),of which approximately 80% were unique to the ERL+CDODA-Me group.Synergistic effects centered on losses to cell cycle progression transcripts,a reduction of minichromosome maintenance complex components(MCM2-7),all key components of the Cdc45·MCM2-7GINS(CMG)complex,and replicative helicases;these effects were tantamount to the upregulation of processes associated with the nuclear factor erythroid 2 like 2 translational response to oxidative stress,including sulfiredoxin 1,heme oxygenase 1,and stress-induced growth inhibitor 1.Collectively,these findings indicate that the synergistic therapeutic effects of ERL+CDODA-Me on resistant NSCLC cells are mediated via the inhibition of mitosis and induction of oxidative stress.
1.Introduction
Lung cancer continues to be a primary cause of mortality worldwide.Erlotinib(ERL)is a standard chemotherapeutic agent for treating patients with lung cancer presenting underlying epidermal growth factor receptor(EGFR)mutations.ERL is a tyrosine kinase inhibitor(TKI)that binds to the ATP-binding pocket of EGFR,thereby inhibiting the protein phosphorylation cascade responsible for driving cell proliferation pathways.Although patients often respond well to TKIs initially,a high rate of relapse and drug resistance has been reported,limiting further treatment[1].It is believed that chemoresistance arises in conjunction with secondary site EGFR and kinase receptor mutations[2,3],activating downstream tumorigenic signaling pathways[4].Accordingly,the identification of effective adjunct chemotherapies is warranted to target divergent adaptive phenotypes through complementary mechanisms.
A combination of multiple TKIs is a proposed strategy to treat resistance;however,this approach has failed clinical trials owing to extensive toxicity[5].As other suitable drug targets could be combined with TKIs,such as growth factor ligands[6],apoptotic signaling[7],inflammation,and angiogenesis[8],we predicted that methyl 2-cyano-3,11-dioxo-18beta-olean-1,12-dien-30-oate(CDODA-Me)would be an ideal adjunct chemotherapy drug to treat ERL-resistant cell lines owing to its diverse properties.CDODA-Me,a glycyrrhetinic acid derivative,has been shown to decrease the expression of specific proteins[9-11];combined with ERL,it causes a synergistic reduction in cell viability[12].In the present study,we evaluated the biological changes in ERL-resistant HCC827R cells when compared with HCC827 controls after treatment with ERL,CDODA-Me,and a combination of both drugs.
2.Materials and methods
2.1.Materials
ERL,tivantinib,dasatinib,osimertinib,olmutinib,crizotinib,and cabozantinib were purchased from MedChemExpress LLC(Princeton,NJ,USA).CDODA-Me was kindly donated by Dr.Stephen Safe(Texas A&M University,College Station,TX,USA).RPMI 1640 medium,fetal bovine serum(FBS),crystal violet,hank balanced salt solution(HBSS),and penicillin-streptomycin-neomycin were purchased from Sigma Aldrich(St.Louis,MO,USA).Primary antibodies were purchased from Cell Signaling Technologies(Danvers,MA,USA),and β-actin was purchased from Santa Cruz Biotechnology,Inc.(Carlsbad,CA,USA).The non-small cell lung cancer cell line,HCC827,was purchased from American Type Culture Collection(ATCC)(Manassas,VA,USA);the HCC827R cell line(ERL-resistant)was provided by Dr.Arun Rishi(Wayne State University,Detroit,MI,USA).
2.2.Cell culture
Cells were incubated at 37°C in 5% CO2and 95% relative humidity,with the medium replaced every two days.Resistant cells were developed according to a previously described method[13],with exposure to increased concentrations of ERL over 12 months,from 0.001 to 4μM.These cells were initially purchased from ATCC and were gifted by Dr.Arun Rishi(Wayne State University,Detroit,MI,USA)after developing ERL resistance[12].
2.3.Cell viability
In brief,cells were seeded in 96-well plates(8,000 cells/well)and incubated at 37°C in 5% CO2for overnight.Afterwards,cells were initially treated with ERL,tivantinib,dasatinib,osimertinib,olmutinib,crizotinib,and cabozantinib for 48 h.Subsequently,cell viability was measured using the crystal violet assay.The combination treatment was assessed by using various concentrations of CDODA-Me(i.e.,producing>75% cell viability)and varying concentrations of TKIs(0.6-100μM).Optimum combinations were selected through isobolographic analysis(Compusyn Software 2.0);then we selected the combination that presented the highest degree of synergism.
2.4.Microarray
After completing the experiments,the cells were scraped,washed three times with ice-cold HBSS,and spun down;then,the supernatant was removed,and the pellet was frozen and stored at-80°C.Total RNA was then isolated and purified using the TRIzol-chloroform method as previously described[14].RNA quality was assessed,and the concentrations were equalized to 82 ng/μL in nuclease-free water.Whole transcriptome(WT)analysis was performed as described in the GeneChipTM WT PLUS Reagent manual for whole transcript expression arrays.Briefly,RNA was reverse-transcribed to first-strand/second-strand cDNA,followed by cRNA amplification and purification.After the second cycle of single-stranded cDNA(ss-cDNA)synthesis and hydrolysis of RNA,ss-cDNA was fragmented,labeled,and hybridized onto arrays prior to fluidics and chip imaging using the Gene Atlas(Affymetrix,Thermo Fisher Scientific,Waltham,MA,USA).Processing of data obtained from the array was performed using expression console software,followed by subsequent analysis using Transcriptome Analysis Console(TAC,Affymetrix,Thermo Fisher Scientific,Waltham,MA,USA)software and STRING database[15].Samples were run in duplicate(n=2).
2.5.Cell cycle analysis
Cells were seeded in 75 cm2flasks(1.5×106cells/flask),allowed to grow overnight at 37°C,and then synchronized for 24 h using 1% FBS-supplemented RPMI 1640 medium.The cells were then reseeded at the same density for treatment with 12μM ERL,2μM CDODAMe,and a combination of the two drugs for 48 h.After treatment,cell pellets were collected according to routine cell culture procedures,re-suspended in 200μL of Dulbecco's phosphate-buffered saline(DPBS),and passed through a 35 mm gauge syringe three times.Next,cells were fixed by adding 70% ethanol in a drop-wise manner under mild vortexing to prevent cell clumping.The cells were then incubated at 4°C overnight for fixation.Fixed cells were centrifuged for 2 min at 2,500 r/min and washed twice with DPBS.Cells were stained for 1 h with a solution containing 50μg/mL of propidium iodide(Cat#P4170;Sigma Aldrich,St.Louis,MO,USA)and 100μg/mL RNase A(Cat#R6513;Millipore Sigma,St.Louis,MO,USA),incubated in the dark at 4°C.Cell cycle analysis was performed using a FACS Calibur flow cytometer(BD Biosciences,San Jose,CA,USA).After the instrument was aligned with Calibrite™beads(BD Biosciences,San Jose,CA,USA),the linearity of the fluorescence pulse was assessed with chicken erythrocyte nuclei(BD Biosciences,San Jose,CA,USA)with a Doublet Discrimination Module according to the manufacturer's protocol.A total of 20,000 cells were acquired using CellQuest Pro software(BD Biosciences,San Jose,CA,USA).Cells in various cycle phases were determined using ModFit LT 3.3.11 software(Verity Software House,Topsham,ME,USA).
2.6.Western blot analysis
Cells were seeded and treated as mentioned above for cell cycle analysis.Protein extraction was performed as previously described using RIPA buffer(Cell Signaling Technologies,Danvers,MA,USA)in a cell lysis cocktail supplemented with phenylmethylsulfonyl fluoride as a protease inhibitor[16].We employed the bicinchoninic acid assay for protein determination according to the manufacturer's instructions.Electrophoresis was performed using 100μg of protein,10% SDS-PAGE gel(Mini-PROTEAN®TGX™Precast Gels),and polyvinylidene fluoride membrane(Bio-Rad Laboratories,Hercules,CA,USA),along with Bio-Rad rapid blot procedures.Membranes were processed priorly by blocking for 3 h with 5% bovine serum albumin,incubated in primary antibody overnight(1:1,000 dilution),and then incubated in horseradish peroxidaseconjugated secondary antibodies for 4 h(1:1,000 dilutions)as described[17].Imaging and protein expression quantification were performed using Clarity™Western ECL Substrate and Image Lab software.
2.7.Statistical analysis
GraphPad Prism 5(GraphPad Software,La Jolla,CA,USA)was used to assess statistical significance.Comparisons were performed using one-way ANOVA,with P<0.05 considered as statistically significant.
3.Results and discussion
3.1.Cell viability
Cell viability studies demonstrated that HCC827 cells were more sensitive to ERL treatment,with an IC50value of 6.43μM when compared with 23.48μM for HCC827R cells.However,HCC827R(ERL-resistant)cells revealed increased tivantinib sensitivity(IC50:8.85 μM)when compared with HCC827 cells(IC50:12.21 μM).Dasatinib sensitivity was greater in HCC827R cells(IC50:6.63μM)than HCC827 cells(IC50:13.35μM).Relevant data are listed in Table 1.

Table 1Cell viability summary showing calculated IC50values following 48 h treatment with various tyrosine kinase inhibitors in both HCC827 and HCC827R cells.

Table 2Cell viability summary showing calculated IC50values following simultaneous 48 h treatment with various tyrosine kinase inhibitors in combination with a constant concentration of 2μM CDODA-Me in both HCC827 and HCC827R cells.
Combination treatment with 2μM CDODA-Me for all TKIs is shown in Table 2,where 2μM CDODA-Me is an adequate concentration to yield the highest degree of synergism[12].The 4μM ERL was selected for treatment in all subsequent assays.
3.2.Pathway analysis
Whole transcriptomic changes for the array sets are presented in Table S1,including control vs.ERL,control vs.CDODA-Me,and control vs.ERL+CDODA-Me(combination),for HCC827R cells.Synergistic effects were observed in the combination group(absent single drug profiles)presented in Table S2.Global summary data for WT differentially expressed genes(DEGs)are presented in Fig.1.In brief,the combination of ERL(4μM)+CDODA-Me(2μM)resulted in 174 upregulated and 210 downregulated genes,comprising a change of 0.36% and 0.44% of the WT,respectively.Among these changes,synergy(absent with individual drugs but present when drugs are combined)was observed in 118 upregulated and 193 downregulated genes,accounting for a considerable percentage of DEGs(91% downregulated DEGs and 68% of upregulated DEGs).

Fig.1.Summary of transcriptome changes in HCC827R cells,treated with either CDODA-Me,ERL or the combination(ERL+CDODA-Me)vs.controls.The data reflect differentially expressed genes(DEGs)from 48,226 transcripts tested as(A)number of DEGs altered and(B)percentage of total transcriptome.CDODA-Me:methyl 2-cyano-3,11-dioxo-18beta-olean-1,12-dien-30-oate;ERL:erlotinib.

Table 3Downregulated genes associated with synergy of the ERL(12 μM)+CDODA-Me(2 μM)co-treatment.The DEG downregulated systems are reflected by STRING database analysis for changes in biological process,molecular function,and cellular components.The data are presented as ID/database,description of system impacted,gene count of DEGs in each network,strength of relationship,and FDR.This table also defines the integrated relational networking by STRING database analysis as presented in the diagram of Fig.2,whereby genes involved(color coded)are presented by symbol in each descriptive network.
Primary synergistic effects of downregulated transcripts unique to the ERL+CDODA-Me group in HCC827R cells were analyzed using the STRING functional association database(Fig.2 and Table 3),confirming the impact on transcripts required to carry out DNA replication.Likewise,using Affymetrix transcriptome analysis,Fig.3 presents the reduced expression of cell division cycle 6(CDC6)and minichromosome maintenance protein complex 2-7(MCM2-7),which would inhibit assembly of the pre-replicative complex and DNA polymerase A binding,ERL+CDODA-Me suppressed MCM10.Furthermore,decreased proliferating cell nuclear antigen(PCNA)expression could cause loss of the RNA-DNA primer production;when combined,this could suppress replicative DNA production required for the cell cycle,also displayed by Affymetrix pathway analysis(Fig.4).
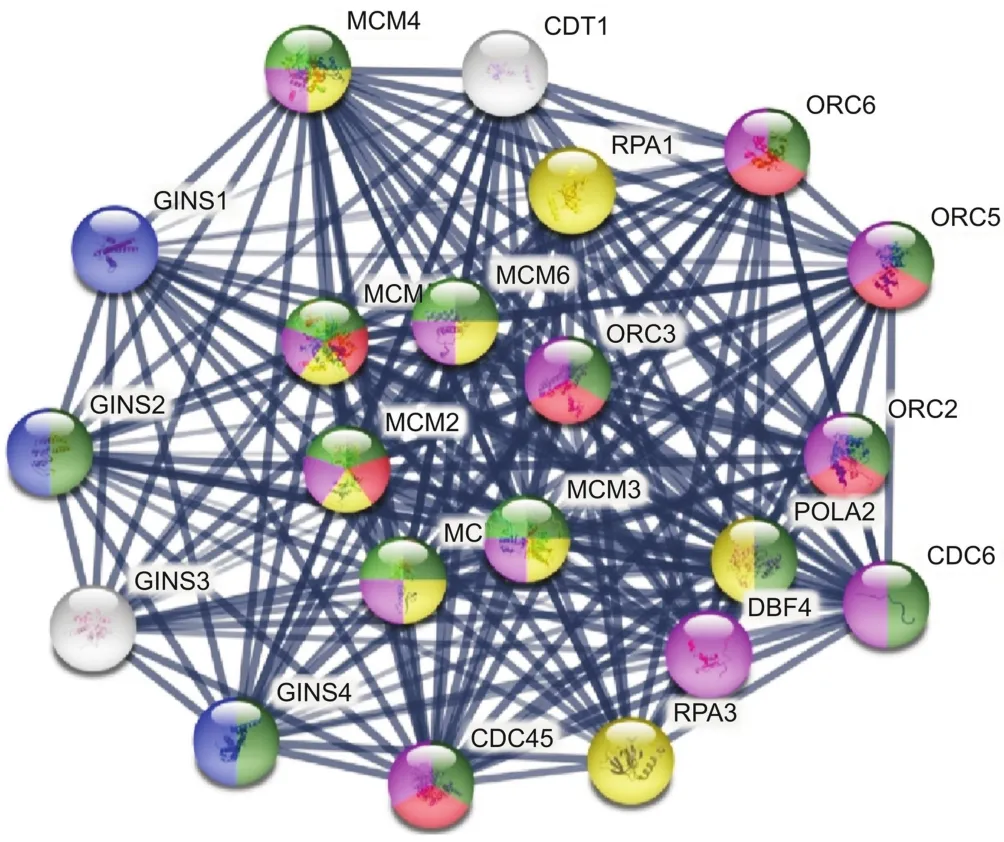
Fig.2.STRING database analysis:synergistic effect of ERL+CDODA-Me on the transcriptome in HCC827R cells.The data were centered on the GINS complex subunit 2 mode to focus on DNA replication and cell cycle.All gene changes are shown in Tables S1 and S2.

Fig.3.DNA replication:ERL+CDODA-Me synergistically affected the DNA replication pathway at 24 h in HCC827R cells.(A)Assembly of pre-replicative complex;(B)activation of pre-replicative complex;(D)DNA replication imitation;(E)leading/lagging strand synthesis.Downregulated transcripts are denoted in solid red.CDC6:cell division cycle 6;CDT1:chromatin licensing and DNA replication factor 1;CDK2:cyclin dependent kinase 2;POLA2:DNA polymerase alpha 2,accessory subunit;PCNA:proliferating cell nuclear antigen.
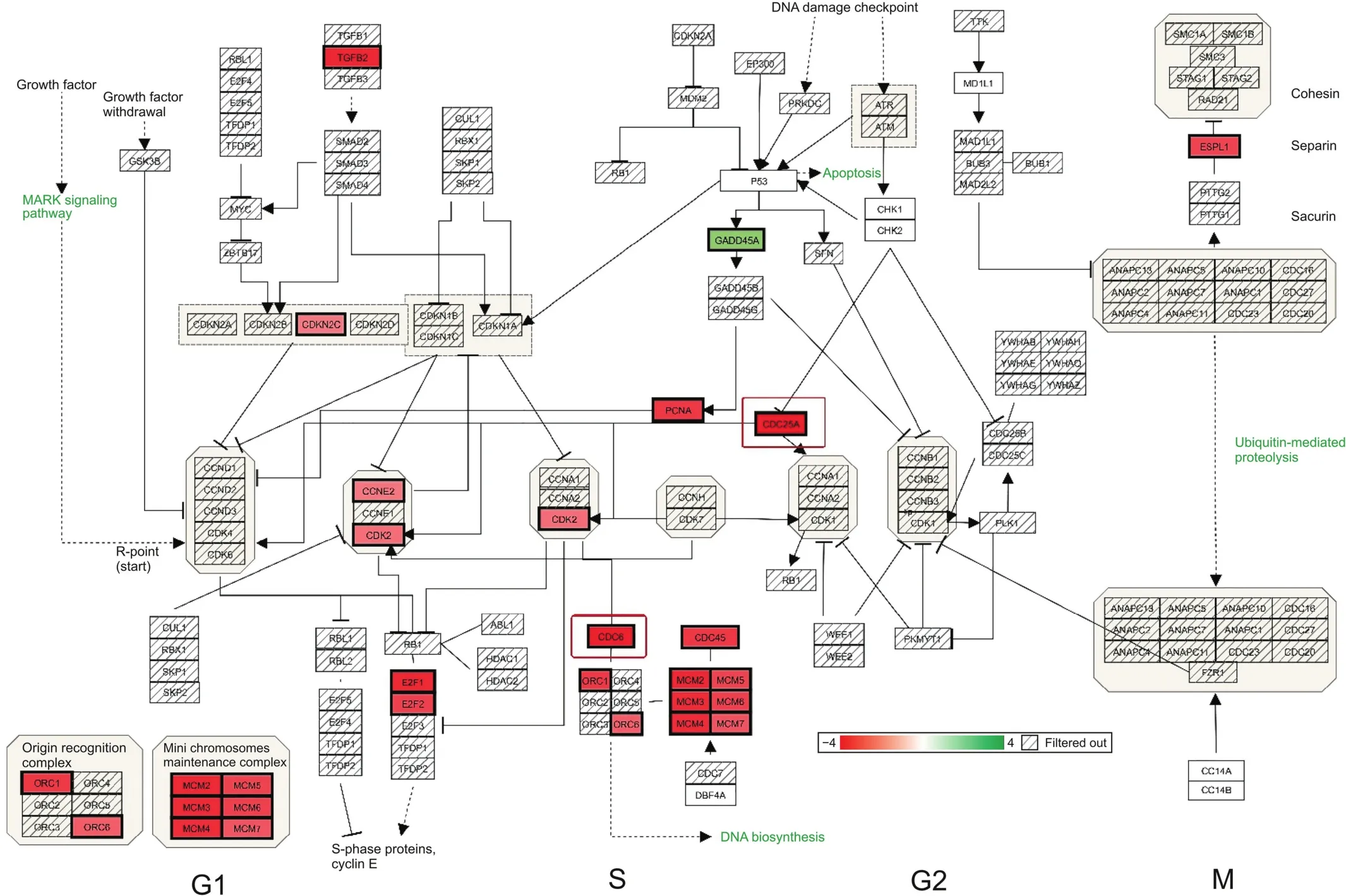
Fig.4.Cell cycle:ERL+CDODA-Me synergistically affected cell division primarily in G1/S in HCC827R cells.Downregulated transcripts are denoted in solid red and upregulated transcripts in solid green.All gene changes are shown in Tables S1 and S2.CCNE2:clycin E2;E2F1:endothelial differentiation-related factor 1;ESPL1:extra spindle pole bodies like 1.
In contrast,synergistic DEGs were upregulated in HCC827R cells,as shown by the STRING functional database(Fig.5 and Table 4),suggesting an oxidative stress component that could aggravate the loss of cell division systems;these effects were also observed in TAC software,indicating upregulation of genes associated with photodynamic therapy-induced nuclear factor erythroid 2 like 2(Nrf2)survival signaling(Fig.6).
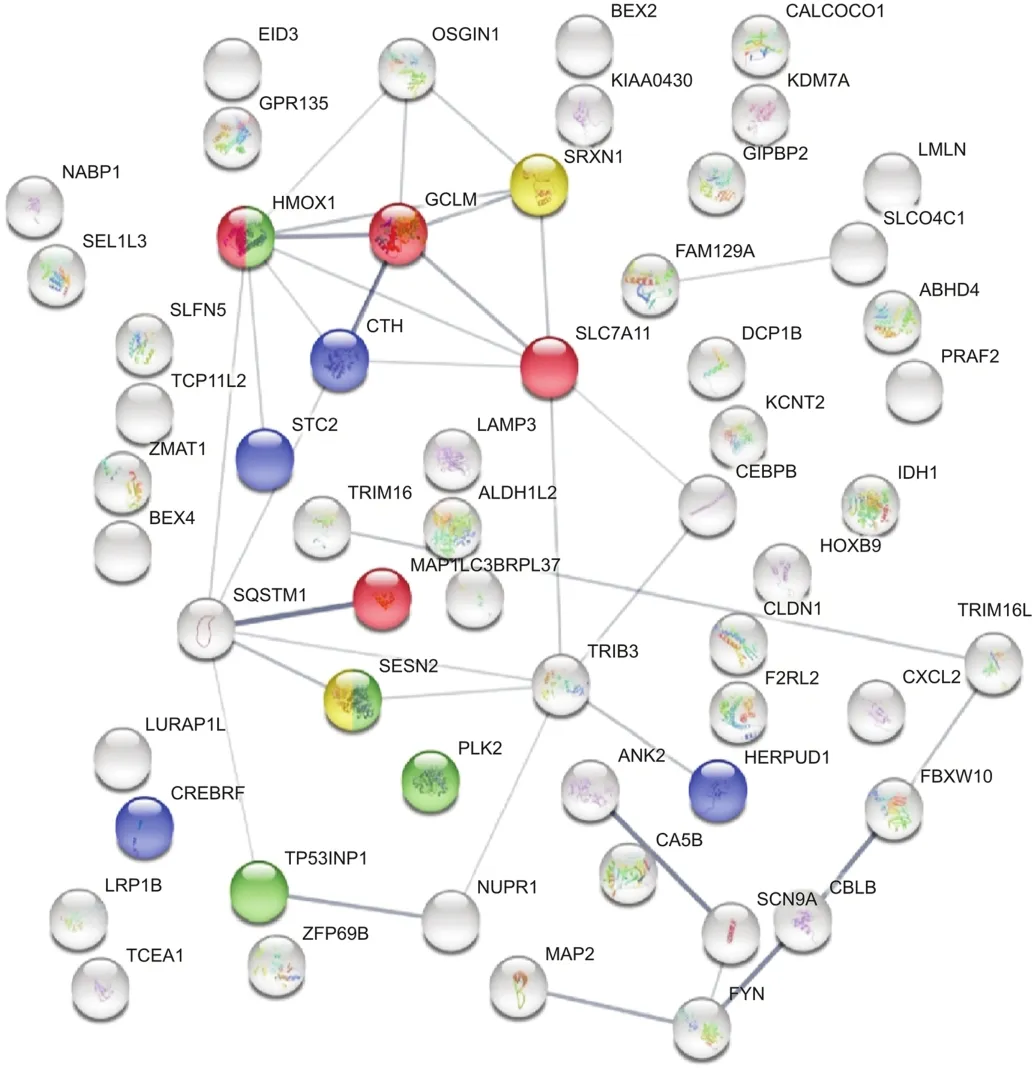
Fig.5.STRING database analysis:synergistic effect of ERL+CDODA-Me on the transcriptome in HCC827R cells.

Fig.6.Photodynamic therapy-induced nuclear factor erythroid 2 like 2(Nrf2)survival signaling:synergistic effect of ERL+CDODA-Me on the transcriptome in HCC827R cells.Upregulated transcripts are denoted by solid green.ROS:reactive oxygen species;HMOX1:heme oxygenase 1;GCLC:glutamate-cysteine ligase catalytic subunit;GCLM:glutamate-cysteine ligase modifier subunit;ABCC3:ATP binding cassette subfamily C member 3;SRXN1:sulfiredoxin 1.
3.3.Cell cycle analysis
To further evaluate the effects of ERL+CDODA-Me on the cell cycle,flow cytometry was performed following treatment with individual drugs and the combination treatment(Fig.7).The data revealed that combination therapy induced cell accumulation in G1 phase,as corroborated by transcriptomic microarray data(Fig.4),where MCM and origin recognition complex were simultaneously downregulated.

Fig.7.(A)Cell cycle analysis(n=3)following treatments:control,CDODA-Me,ERL,and 12μM ERL and 2μM CDODA-Me in HCC827R cells.Cell pellets were collected following normal cell culture procedures,fixed with 70% ethanol and stained with propidium iodide before measuring using flow cytometry.***Compared to control(untreated cells),the significance is defined as P<0.001.(B)Flow cytometry data charts.
3.4.Western blot analysis
Western blotting was employed to evaluate several proteins involved in the cell cycle and revealed that the drug combination significantly decreased protein expressions of CDC25A,CDC6,and thymidine kinase,consistent with mRNA changes in the transcriptome(Fig.8 and Table 5),further corroborating central loss to DNA replicative cell cycle components.

Table 5Downregulated gene expression changes for CDC25A,thymidine kinase 1(TK1),and CDC6 reflecting synergy of the ERL(12μM)+CDODA-Me(2μM)co-treatment.The data represent expression by average log2,fold change,P value and FDR P value.

Fig.8.Western blotting and densitometric analysis of investigated proteins(n=3).Control,CDODA-Me,ERL,and 12 μM ERL and 2 μM CDODA-Me combination in HCC827R cells.Total protein expression of CDC25A,CDC6,and thymidine kinase after 48 h of treatment,relative toβ-actin expression are presented.***Compared to control(untreated cells),the significance is defined as P<0.001.

Table 4Upregulated genes associated with synergy of the ERL(12μM)+CDODA-Me(2μM)co-treatment.The DEG upregulated systems are reflected by STRING database analysis for changes in biological process and molecular functions.The data are presented as ID/database,description of system impacted,gene count of DEGs in each network,strength of relationship,and FDR.This table also defines the integrated relational networking by STRING database analysis as presented in Fig.5,whereby genes involved(color coded)are presented by gene symbol in each descriptive network.
4.Discussion
Chemotherapeutic agents for lung cancer include EGFR TKIs such as ERL[1].Given the inherent propensity for relapse and chemoresistance to ERL,combination drug strategies remain essential[18].We have previously reported combination therapies to overcome drug resistance by increasing the potency of ERL at the tumor site by enhancing bioavailability[19],tumor targeting[20,21],and tumor permeation[22].In the present study,we performed WT analysis to identify changes in mRNA,to assess the potential anticancer mechanism of action underlying CDODA-Me when combined with ERL in HCC827R cells.
Specifically,we will discuss only those changes exclusive to the ERL+CDODA-Me transcriptomic profile(neither occurring in the ERL or CDODA-Me treatments individually when compared with controls).In the present study,functional pathway analysis revealed that the drug combination negatively impacted cell division and chromosomal replication processes.Differential gene expression patterns revealed downregulation of transcripts for all six MCM components(MCM2-7),in addition to all key components of the DNA replication CDC45/MCM2-7/GINS(CMG)replicative helicase complex.The integrity of the CMG complex is essential for replicative DNA helicase activity,as it interacts with topoisomerase at DNA replication forks[23]to maintain leading strand synthesis[24].The synergistic loss of the aforementioned component,in addition to the GINS complex subunit 2(Psf2 homolog,GINS2),can consolidate anti-mitotic effects owing to its vital role at eukaryotic replicative forks for initiation of DNA replication[25].Overexpressed GINS-related genes are involved in several types of cancers associated with aggressive malignant tumors,including breast,melanoma,liver,and lung cancers[26].In lung cancer,the overexpression of GINS2 transcripts correlates with rapid tumor growth;GINS2 knockdown reportedly attenuates several aspects of tumor growth,including tumor size[27,28],migration,invasion[29],stem-like phenotype transition[30],and cell proliferation,owing to loss of DNA replication required for the G2/M phase[31].
In addition to the six downregulated transcripts(MCM2-7)of the CMG complex and GINS2,ERL+CDODA-Me suppressed MCM10,which is a critical target owing to its essential role in the activation of CMG complex helicase activity[32],initiation of eukaryotic chromosomal DNA replication,and binding of the helicase complex to chromatin to stabilize the collective attachment of CDC45 and GINS association with MCM2-7[33].Synergistic effects of the two drugs inhibited CDC45,required in the CMG complex to unwind duplex DNA[34].The loss of MCM10 is pivotal given its indispensable role in DNA replication,strand elongation[35-38],replication fork stability,recruitment,and binding of DNA polymerases and PCNA[39].A collective loss in this area of the transcriptome reinforces the collapse of the replicative helicase machinery required for DNA replication and cell proliferation[40].MCM10 is highly overexpressed in diverse human cancers and is a prognostic indicator of poor overall survival[41,42],advanced clinical stage,and high Gleason score[43,44].As expected,experimental knockout of MCM10 induces cytostatic effects,reduces tumor formation and migration capacity[45],and promotes the anchorage-independent growth of tumor cells[46].Numerous other related transcripts involved in DNA replication and cell cycle were also downregulated by ERL+CDODA-Me,including PCNA,flap endonuclease 1,CDC6,CDC25A,and histone cluster 1,all required for cytokinesis and cell proliferation in various tumors,including lung cancer[47-50].Spot testing of several expressed proteins,found to be reduced at the mRNA transcript levels,was corroborated by Western blotting,indicating a loss of CDC25A,CDC6,and thymidine kinase.The synergistic drug effects observed at the transcriptomic levels were also detected biologically by arresting the cell cycle,as elucidated by flow cytometry,at the G1 phase,which has been previously reported[51-53].
The second synergistic effect of ERL+CDODA-Me was indicative of oxidative stress events such as ferroptosis,which is typically associated with the upregulation or downregulation of iron-related transcripts,such as heme oxygenase-1[54,55],ferritin heavy chain 1(FTH1)[56],transferrin receptor 1,ferroportin,and altered expression of glutathione-related genes such as cystine/glutamate transporter xc(-)(SLC7A11),glutathione peroxidase 4(GPX4),and glutamate-cysteine ligase,which are regulated by Nrf2 transcription in response to oxidative stress.ERL+CDODA-Me results in upregulation in Nrf2,similar to that observed as a survival response triggered by the presence of oxidative dangers by elevating intracellular glutathione-related antioxidant systems[57].Drugs that act as iron chelators or ferroptosis inhibitors[58,59]can inhibit cell death,while inhibition of cysteine uptake,attenuation of glutathione transferases,or inactivation of GPX4 can exacerbate it[60].Increased levels of heme oxygenase,as induced by ERL+CDODAMe,are often observed in response to reactive oxygen species,degrading heme into biliverdin,both of which afford protection against stress;simultaneously,it can release ferrous iron,which could act as a pro-oxidant.Accordingly,heme oxygenase is a critical double-edged element that could afford protection or trigger cell death[60].Future research is needed to investigate this aspect of the synergism between ERL+CDODA-Me and oxidative stress.
5.Conclusions
The above findings represent the WT data profile of ERL and CDODA-Me combination in ERL-resistant lung cancer cells.Combining these two drugs presented synergistic negative effects on DNA replication and cell proliferation while promoting oxidative stress.This work adds to our existing research in exploring potential chemo sensitizing agents in drug resistant tumor models[12,61-63].
Declaration of competing interest
The authors declare that there are no conflicts of interest.
Acknowledgments
We thank National Institute on Minority Health and Health Disparities(National Institutes of Health,Grant/Award No.:U54 MD007582)and NSF-CREST Center for Complex Materials Design for Multidimensional Additive Processing(CoManD,Grant/Award No.:1735968)for providing the funding for this research.
Appendix A.Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jpha.2021.06.002.
杂志排行

Journal of Pharmaceutical Analysis的其它文章
- Evaluation of the gastrointestinal anti-motility effect of Anacardium occidentale stem bark extract:A mechanistic study of antidiarrheal activity
- Simultaneous enantioseparation and simulation studies of atenolol,metoprolol and propranolol on Chiralpak®IG column using supercritical fluid chromatography
- Spectroscopic studies of the interaction between phosphorus heterocycles and cytochrome P450
- Impaired tricarboxylic acid cycle flux and mitochondrial aerobic respiration during isoproterenol induced myocardial ischemia is rescued by bilobalide
- Novel insights into conjugation of antitumor-active unsymmetrical bisacridine C-2028 with glutathione:Characteristics of non-enzymatic and glutathione S-transferase-mediated reactions☆
- A living cell-based fluorescent reporter for high-throughput screening of anti-tumor drugs