分子氧在Con小团簇吸附的密度泛函研究
2022-01-19颉雨佳
颉雨佳 袁 佩
刘 叶1 朱裔荣1
1.湖南工业大学
材料与先进制造学院
湖南 株洲 412007
2.湘潭大学
化工学院
湖南 湘潭 411105
1 研究背景
环己烷中C—H键的键能较强(376.81~418.68 kJ/mol),在化学反应中难氧化,且产物KA油(环己醇和环己酮)选择性差,在有氧体系中易于过度氧化生成酸或酯。目前,工业上常采用钴盐或锰盐体系对环己烷进行催化反应,但此类反应转化率较低且存在后续催化剂较难分离回收等环保问题[1]。
在解决环己烷催化反应难题时,分子氧和金属钴之间的活性及稳定性是表面氧化、表面腐蚀及多相催化过程中至关重要的影响因素[2]。目前,国内外研究主要集中于Co体系对环己烷催化的机理研究。Zhang P.等[3]选用钴基尖晶石纳米晶作为催化剂(LP@ZIF-67),在O2压力为2.0 MPa时,得到催化环己烷的转化率为17.2%,KA油的选择性为95.3%。Shen H. M. 等[1]采用卟啉铜协同卟啉钴催化环己烷,KA油的选择性从88.6%提高到97.2%,转化率从3.88%提高到4.41%。Hao F. 等[4]采用N掺杂石墨烯负载的钴催化剂(Co-N-rGO)催化环己烷,得到环己烷转化率为8.85%,KA油选择性为85.73%。N. Shahzad等[5]采用 VASP软件进行模拟计算,发现Co团簇的吸附对团簇尺寸和表面结构非常敏感,且团簇在原始石墨烯表面的成核比缺陷石墨烯表面的成核更有利。K. García-Díez 等[6]采用Dacapo软件进行理论计算,结果表明H2在Co6和Co13团簇的吸附和解离表现出相似的特征,但Co6团簇理想储氢量达到 8.4%。Li T. T. 等[7]采用 VASP 软件进行模拟计算,结果表明O2和CO分子在最高对称性的Cu12Ni团簇上的吸附比在Cu13和Ni13团簇上更适合CO氧化。
依据绿色化学反应原理,直接利用O2将分子氧或原子氧插入烷烃中的C—H键或C—C键中,活化的分子氧进一步高效活化环己烷,氧化生成中间体(环己基过氧化氢),进一步生成KA油。这不仅有利于反应以最短路径生成相应的重要含氧化合物[8-9],而且具有原子经济性高的特点。因此,本研究通过密度泛函计算钴团簇活化分子氧的最优结构,系统研究Con(n=1~5)团簇和Con团簇对分子氧垂直(V(⊥))和平行(P(∥))吸附的几何结构(ConO2)、稳定性和电子性质,并对相关能量体系进行计算,计算模拟结果可为吸附、防腐蚀和催化应用(CO氧化、环己烷和烷烃氧化)实验研究提供理论指导,对无碳、可持续的替代能源技术具有重要意义。
2 计算方法
采用Dmol3程序包进行密度泛函理论计算。所有结果均由广义梯度近似(generalized gradient approximation,GGA) 的 Perdew-Burke-Ernzerhof(PBE)泛函得到。在几何优化中,所有的Con团簇结构都是保持弛豫,对称性不限制、自旋限制,计算过程中核处理采用全电子方式,基组为双数极化(double-numerial basis,DNP)。精度选择fine,能量和位移分别设置为 1 × 10-5Ha 和 0.005 Å,直到收敛。为了验证所选方法和参数的可靠性,在该条件计算O2的键长为1.227 Å,与实验值1.207 Å[2]相比误差较小,由此表明该方法和参数的可靠性。
通过文献调研并对 Con(n = 1~5)团簇结构进行优化,获得具有最低能量的结构。优化的Con团簇顶位(top,T)、桥位(bridge,B)、空位(hole,H)的位置如图1所示。
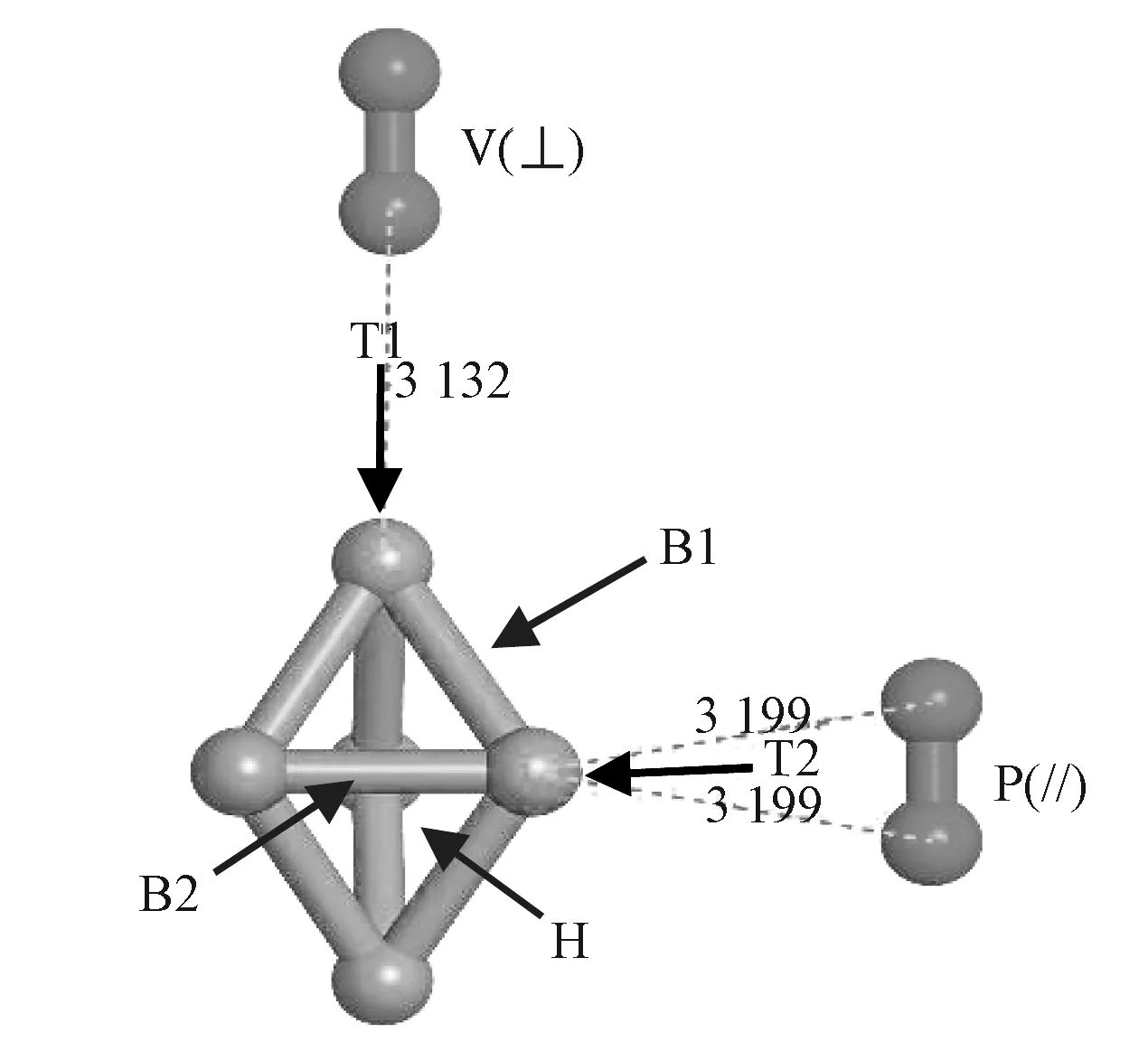
图1 分子氧在团簇上的吸附结构Fig. 1 Adsorption structure of molecular oxygen on clusters
分子氧预置方式有:垂直活性位点(V(⊥))和平行活性位点(P(∥))。在初始垂直距离大于3 Å的位置预置分子氧,在相同的条件下进行结构优化,对优化后结构进行频率计算无虚频,以确保结构是最低能量的稳定结构[10],之后进行性质的计算。
团簇 Con(n = 1~5)的结合能定义:
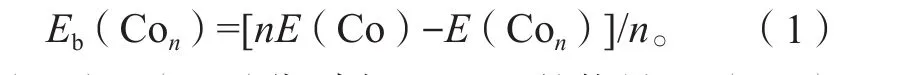
其中:E(Co)、E(Con)分别为Co、Con的能量;Eb(Con)为结合后的总能量。结合能越大,结构越稳定。
团簇ConO2(n=1~5)的结合能定义:
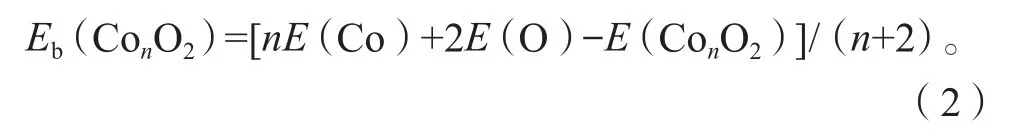
其中:E(Co)、E(O)和E(ConO2)分别为Co、O和ConO2的能量;Eb(ConO2)为结合后的总能量。结合能越大,结构越稳定。
分子氧(O2)在团簇上的吸附能定义:

其中:E(ConO2)为吸附分子氧后的总能量;E(O2)为O2的能量。吸附能Ea为负值,表示吸附放热,绝对值越大,吸附越强。
3 结果与讨论
3.1 Con(n = 1~5)团簇的稳定几何结构
图 2 是经过优化后的 Con(n = 1~5)团簇的几何结构图。
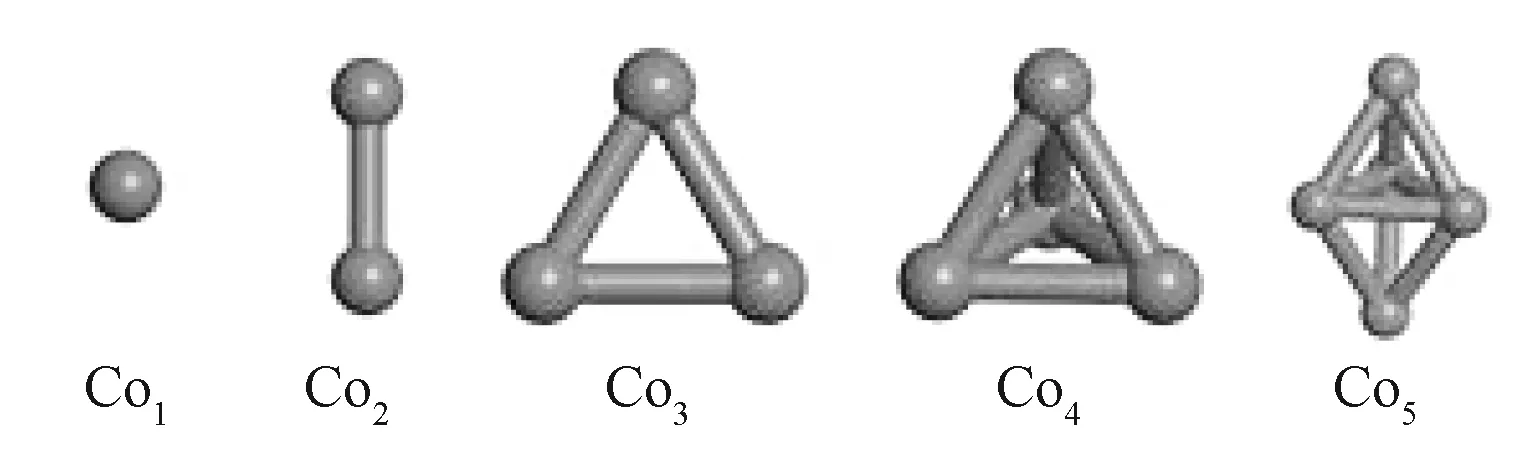
图2 Con团簇优化后的几何结构Fig. 2 The optimized geometries of Con clusters
能量最高的最高占据分子轨道(highest occupied molecular orbital,HOMO)可提供电子成键,而能量最低的最低未占据分子轨道(lowest unoccupied molecular orbital,LUMO)可接受更多的电子,带隙能(Gap)是LUMO与HOMO的差值,较大的带隙能意味着较高的动力学稳定性和较低的化学反应活性[11]。带隙能反映电子被激发的难易程度和所需能量大小,其值越大,表示该分子越难以激发,活性越差。Con(n = 1~5)团簇的结合能(按式(1)计算)和带隙能如表 1 所示。Con(n = 2~5)团簇的结合能随团簇数目的增加而增大,变化趋势趋于平缓,表明团簇的稳定性随团簇尺寸的增加逐渐增强。随原子数目的增加,Con团簇的Gap整体变小,说明动力学上参与化学反应的活性显著增强。在n=2~4的条件下,可以观察到团簇的奇偶振荡效应[12]。
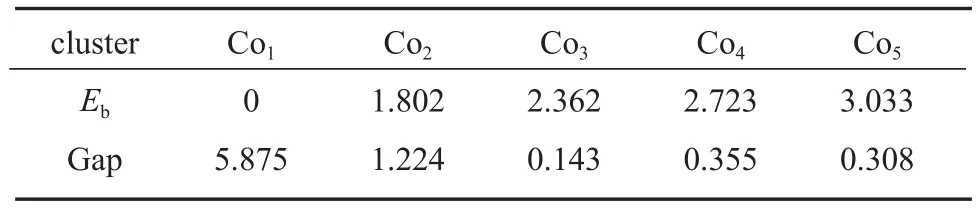
表1 Con(n=1~5)团簇的结合能和带隙能Table1 Binding energies and gap energies of Con(n=1~5) clusters eV
3.2 ConO2(n=1~5)团簇的稳定性分析
研究Con团簇吸附分子氧后的电荷转移和化学活性,需考察分子氧在Con团簇上不同的吸附位点:顶位、桥位和空位的吸附。初始分子氧预置的吸附方式有:垂直吸附(V(⊥))和平行吸附(P(∥))。垂直和平行吸附优化后的构型如图3所示。

图3 优化所得ConO2团簇几何结构Fig. 3 The optimized geometries of ConO2 clusters
对比图3a中ConO2团簇几何结构可以发现,初始分子氧垂直活性位点的结构经过几何优化后,除了Co3O2H和Co5O2T2团簇基本保持分子氧垂直活性位点外,其余结构都有明显偏移。Co5O2H团簇中Co5团簇结构由三角双锥转变成了四角锥;Co2O2B、Co5O2B1、Co5O2B2 和 Co5O2H 团簇中分子氧的键长显著伸展。图3b中初始分子氧平行活性位点的结构经过几何优化后,整体上依然保持平行活性位点的构型。Co4O2H团簇中 Co4团簇有些变形;Co5O2B2和Co5O2H团簇中Co5团簇也由三角双锥转变成了四角锥;Co2O2B、Co3O2H、Co4O2H 和 Co5O2H 团簇中的分子氧键长也显著伸展。
分子氧在Con团簇不同位置垂直和平行吸附后,根据式(3)和式(2)计算所得ConO2团簇的吸附能和结合能如图4所示。
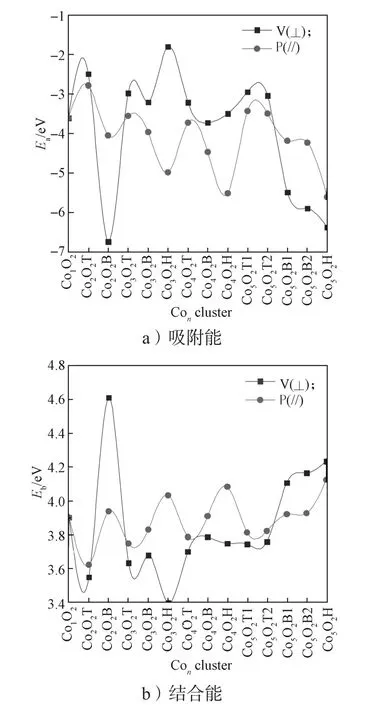
图4 ConO2团簇的吸附能与结合能Fig. 4 Adsorption energy and binding energy of ConO2 clusters
由图4可知,优化后的吸附能和结合能变化趋势一致,分子氧垂直吸附的吸附能和结合能最强是Co2O2B 团簇,分别是 -6.744 eV 和 4.611 eV;最弱的是 Co3O2H 团簇,分别是 -1.806 eV 和 3.398 eV。分子氧垂直吸附型 Co2O2B、Co5O2B1、Co5O2B2 和Co5O2H团簇的吸附能和结合能都比同类型平行吸附的要大,而其余结构则相反。分子氧平行吸附的团簇中,同数目不同位置的吸附能和结合能由大到小依次是:E空位>E桥位>E顶位。负值表明吸附放热,绝对值越大,吸附越强;结合能越大,说明分子氧吸附后的结构越稳定。对比相应优化的几何结构,分子氧键长没有明显伸长。
ConO2团簇中Co—O最短键长随团簇的变化如图5所示。由Co—O的最短键长与ConO2团簇数目和位置的关系表明,键长(d)介于 1.600 ~ 1.988 Å之间,Co—O的键长都显著比初始设定距离(3 Å)要小。在分子氧垂直吸附构型的团簇中, n≤4时同数目不同位置的Co—O最短键长由大到小依次是:d空位>d桥位>d顶位;而n=5时,顶位上的键长大于桥位、空位的。在分子氧平行吸附构型的团簇中,同数目不同位置的Co—O最短键长由大到小依次是:d顶位> d桥位>d空位。

图5 ConO2团簇上Co—O的键长Fig. 5 Bond length of Co—O on ConO2 clusters
ConO2团簇中O—O键长随团簇数目和位置的变化如图6所示。
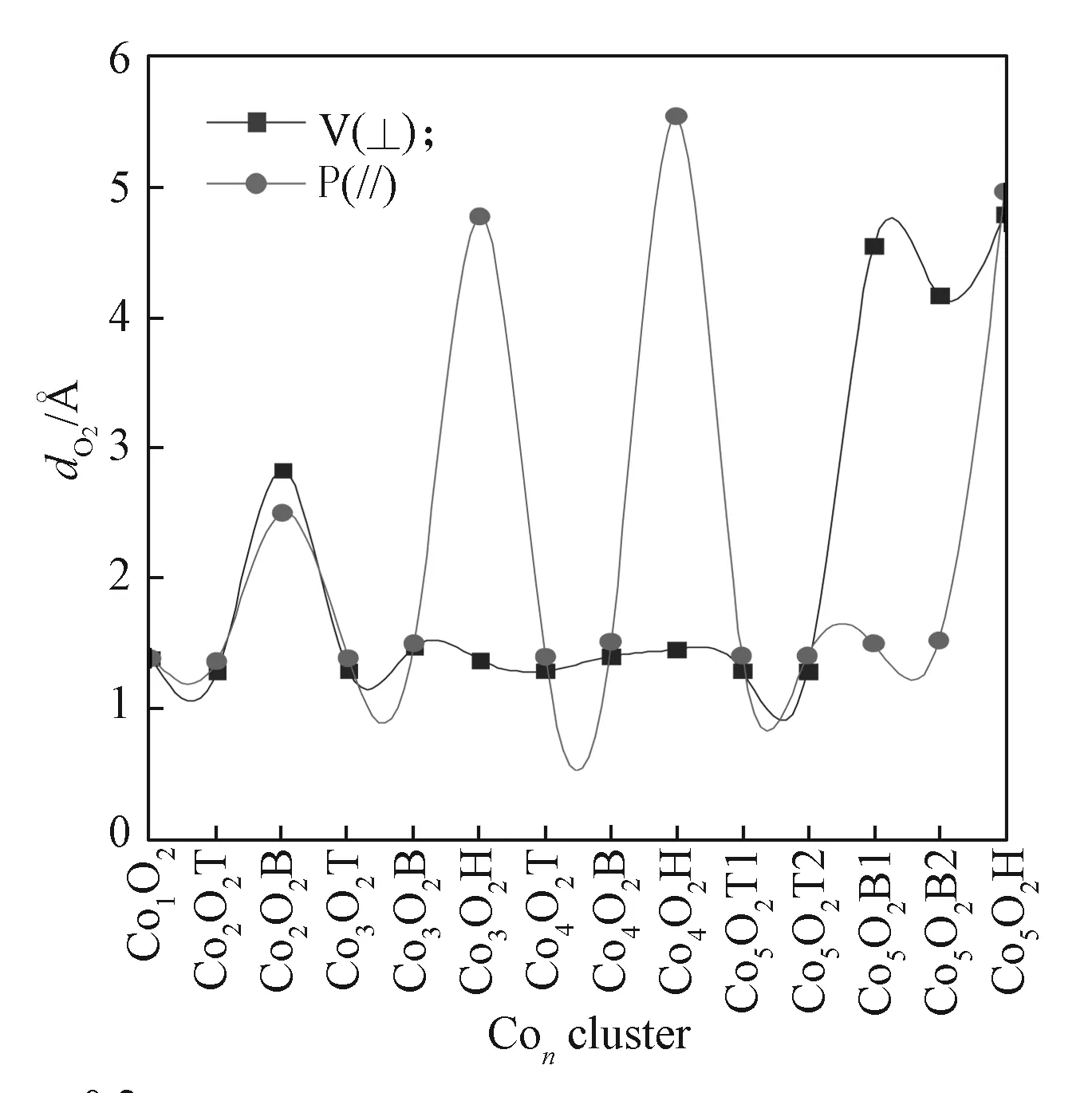
图6 ConO2团簇上O—O的键长Fig. 6 Bond length of O—O on ConO2 clusters
由图6可知,初始O2垂直构型的Co2O2B、Co5O2B1、Co5O2B2、Co5O2H 和平行构型的Co2O2B、Co3O2H、Co4O2H、Co5O2H 团簇,O—O 键长介于2.498 ~ 5.543 Å,偏向于裂解;其余结构中的O—O键长主要介于 1.282 ~ 1.518 Å,但都比自由态 O2的O—O键长(1.227 Å)要长。在几何结构上,ConO2团簇中的Co—O的键长都显著比初始设定距离(3 Å)要小,介于1.600 ~ 1.988 Å之间,进一步表明了Con团簇对分子氧的活化作用。
3.3 ConO2(n=1~5)团簇的电子性质分析
团簇的稳定性和反应活性与电子结构密切相关。由于原子氧(O)的电负性远大于原子钴(Co)的,故分子氧的得电子能力较强。因此团簇结构吸附分子氧之后,ConO2团簇中分子氧因得电子而带负电,分子氧中的原子氧所得的电荷,随着原子氧与同一原子钴距离的增长而增大;金属团簇因失电子而带正电。电荷转移进一步表明团簇活化了分子氧[13]。
采用Mulliken电荷分析团簇和分子氧之间的电荷转移,两者间电荷变化情况如图7所示。由图7可知,相同数目n、不同位置上O2得电子的能力由强到弱依次是:空位、桥位、顶位;O—O键长变化小的得电荷介于 -0.281 ~-0.611 e;O—O 键长变化大的得电荷介于 -0.814 ~-1.130 e。这是由于 Con团簇电子向分子氧反键π轨道的转移,导致电荷在两个O上分布不均和Co—O键不相等,O—O键强度削弱而伸长。Con团簇和分子氧之间的电荷转移越多,相互作用越强,吸附能和结合能也越大。
图7 ConO2 团簇上的 O2 电荷分析Fig.7 Charge analysis for O2 on ConO2 clusters
ConO2团簇的带隙能如图8所示。由图8可以看出,ConO2团簇的Gap变化规律性不强,Gap值介于0.031~0.535 eV;这主要是因为O2吸附到Con团簇后的带隙能不仅与分子氧的吸附方式有关,而且与吸附的位置有关,协同作用的结果直接影响吸附后团簇的化学活性。分子氧为强电子接受体,根据吸附后得电荷的情况,主要提供LUMO轨道,接受Con团簇HOMO轨道上的电子,吸附的强弱与轨道的电子云密度高低直接相关。
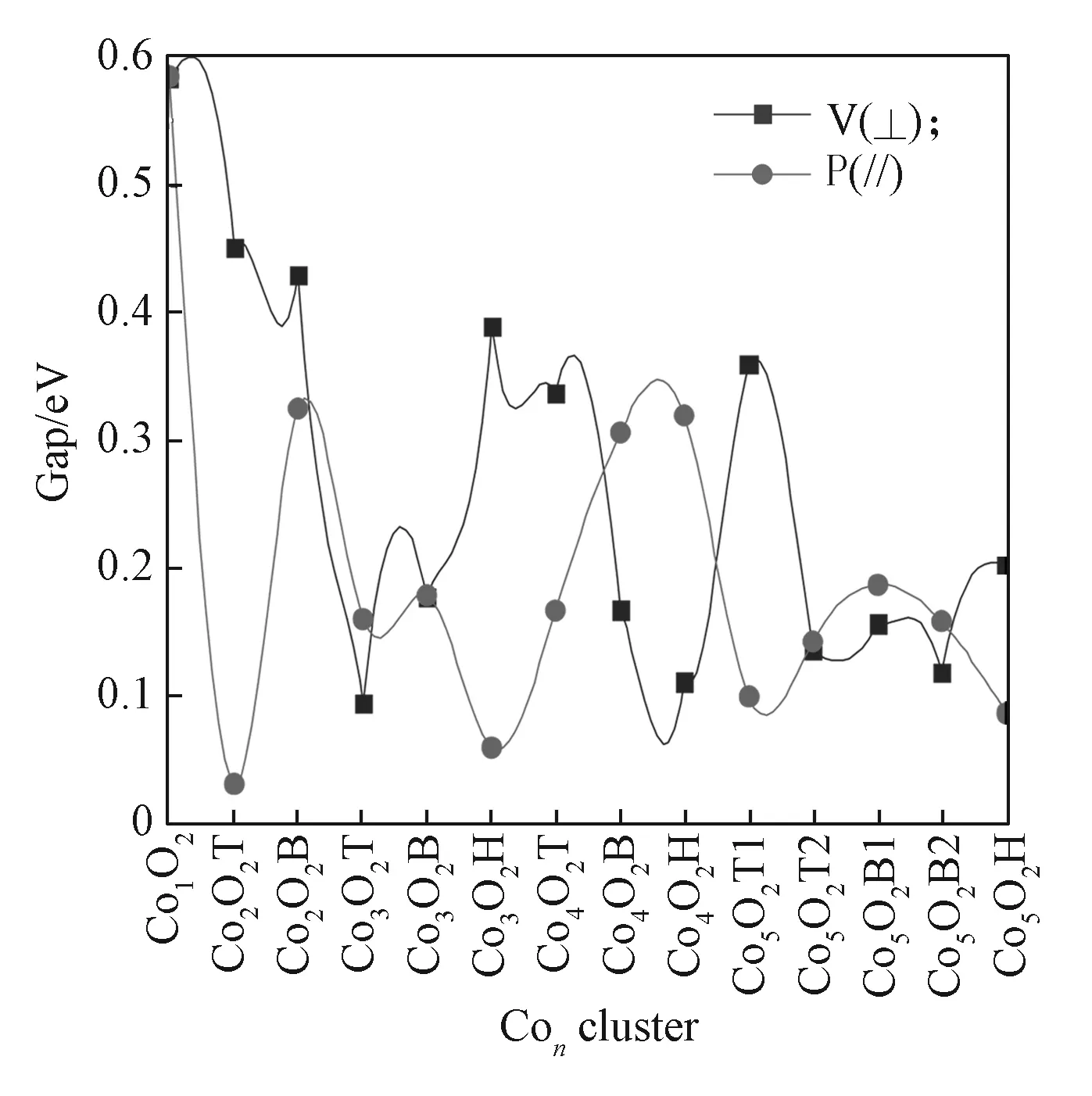
图8 ConO2团簇的带隙能Fig. 8 Gap energies of ConO2 clusters
4 结论
采用密度泛函理论系统研究Con团簇对分子氧垂直和平行吸附的几何结构(ConO2)、稳定性和电子性质的影响,并对相关能量体系进行计算,得出的主要结论如下。
1)从吸附能和结合能可看出,分子氧垂直吸附的吸附能和结合能最强的是Co2O2B团簇,分别是 -6.744 eV 和 4.611 eV;最弱的是 Co3O2H 团簇,分别是 -1.806 eV 和 3.398 eV。
2)ConO2团簇中部分 O—O 键长介于 2.498 ~5.543 Å,偏向于裂解;其余的O—O 键长主要介于1.282 ~ 1.518 Å,总体上 O2键长变化都比自由态 O2的O—O键长(1.227 Å)要长,净增长为0.055 ~ 4.316 Å。Con团簇向分子氧反键π轨道转移电子,导致电荷在两个O上分布不均和Co—O键不相等,O—O键强度削弱而伸长。
3)在Mulliken电荷转移上,ConO2团簇中分子氧得电子带负电,金属团簇失电子带正电,O—O键长变化小的得电荷介于 -0.281 ~ -0.611 e;O—O 键长变化大的得电荷介于-0.814 ~-1.130 e。键长和电荷转移进一步表明Con团簇活化了分子氧。
