水热碳化法制备淀粉碳化物/海泡石(St-Sep)复合材料的优化
2022-01-17郑锡瀚马忻狄潘怡莹刘雪英蓝丽红
郑锡瀚,马忻狄,潘怡莹,刘雪英,蓝丽红
(广西民族大学化学化工学院,广西多糖材料与改性重点实验室,广西高校化学与生物转化过程新技术重点实验室,广西 南宁 530006)
海泡石是由金属元素阳离子与硅酸根结合的层链结构纤维状的含水镁硅酸盐矿物[1],其理论化学式为Mg8[Si12O30](OH)4·12H2O[2]。天然海泡石以纤维状存在,其内部孔隙多且孔径较大,使其具有较大的比表面积及良好的吸附性能[2-3]。由于海泡石内部含有较多的结晶水和吸附水,造成了其吸附容量减小[4]。此外,在我国乃至全球范围内,海泡石矿产储量丰富且年开采量极大,却大多以原料进出口的形式进行廉价贸易[5]。
近年来通过物理和化学方法改性提高海泡石吸附性能已成为矿物加工和应用领域的研究热点。其中主要改性方法有酸改性[6]、有机改性[7]、表面涂层增白处理[8]和连续还原法[9]等。然而,无机溶剂和有机溶剂改性都会造成二次污染,且溶剂难以回收。水热碳化法[10-12]制备海泡石基复合材料的反应条件更为温和可控、能耗低、经济绿色,并且糖类物质在较高温度和压力的条件下,会发生脱水、聚合和碳化反应,形成易与其他材料发生结合作用且存在多种活性官能团(如,C-H、C=O和C=C等)[13]的不完全碳化的糖类碳化物[14]。通过在海泡石表面负载糖类碳化物,引入大量活性官能团,以达到增强材料表面的吸附功能,在很大程度上拓展了海泡石基复合材料的应用领域。
有关葡萄糖[15-16]、果糖[17]、纤维素[18]和淀粉[19-22]等生物糖类物质的水热碳化反应的研究在国内外均有见报导。相对而言,淀粉是仅次于纤维素的第二大生物多糖,广西地区产量较大。因此,以海泡石为载体,广西木薯淀粉为改性剂,利用水热碳化法,制备St-Sep复合材料,并使用冷冻真空干燥法干燥复合样品。并用X射线衍射分析(XRD)、红外吸收光谱分析(FTIR)、扫描电镜(SEM)和比表面积仪(BET)对复合材料进行表征。选择亚甲基蓝作为吸附质,以St-Sep复合材料对亚甲基蓝的吸附量为性能考察标准,通过单因素和响应面法优化了材料的制备工艺。旨在获得一种吸附性能优良的复合型海泡石基材料,提高海泡石的附加值的同时,又能为地区丰富的多糖类物质利用开辟新的应用途径,为海泡石和多糖类物质的深加工及应用提供实验依据和较好的技术指导。
1 材料与方法
1.1 材料及仪器
海泡石(Aldrich);木薯淀粉(工业级);六水合硫酸亚铁铵(分析纯); 亚甲基蓝(Solarbio,含量为98%~103%,MB)。
MAGNA-IR550型傅立叶变换红外光谱仪;SUPRA 55 Sapphire型场发射扫描电子显微镜;Rigaku miniflex 600型 X射线衍射仪;ASAP2460型比表面积测定仪;TU-1810PC型紫外可见光分光光度计。
1.2 实验方法
1.2.1 淀粉碳化物/海泡石复合材料的制备
将4.5 g木薯淀粉溶于蒸馏水中,再将0.5 %六水合硫酸亚铁铵(催化剂)[19]加入淀粉中,待分散之后,按比例(淀粉与海泡石的添加质量比)添加海泡石,与35 mL蒸馏水混合,配置成混合悬浊液。搅拌30 min,再将悬浮液超声(超声频率为40 kHz)分散30 min。最后将混合样品转移至50 mL不锈钢反应釜的聚四氟乙烯内衬中(实验过程中保持样品量占釜内体积约为80%,主要为了控制反应釜内部压强等条件一致),180℃下进行水热碳化反应8 h,所得样品用无水乙醇与蒸馏水交替洗涤,直至滤液为无色[23]。将粗样品进行真空冷冻干燥后,即可得到St-Sep复合材料。
1.2.2 淀粉碳化物/海泡石复合材料的吸附性能探究
准确称取0.1020 g亚甲基蓝(MB,实验用亚甲基蓝含量为98%~103%),用蒸馏水配制成浓度为100 μg/mL的亚甲基蓝原液,并进一步稀释,分别得到浓度分别为0、1.0、2.5、5.0、7.5、10 μg/mL的系列浓度梯度的亚甲基蓝溶液,以蒸馏水作为参比,在662 nm处测定亚甲基蓝系列溶液的吸光度,并绘制标准曲线。
精确移取50 mL亚甲基蓝原液于150 mL的具塞锥形瓶中,加入0.110 g复合材料样品,室温条件下振荡4 h吸附后,于10000 r/min条件下离心分离,吸取上清液测定吸光度,并计算海泡石对亚甲基蓝的吸附量。
在本次实验中,吸附量和吸附率分别采用式(1)、(2)进行计算[24]:

式中:Q为吸附量,mg/g;R为吸附率,%;C0为亚甲基蓝溶液初始浓度,μg/mL;Ct为吸附完成的剩余液浓度,μg/mL;V为添加溶液体积,mL;M为复合材料投加量,g。
1.2.3 表征方法
1.2.3.1 扫描电镜(SEM)
实验样品干燥后,取少量黏附于导电胶之上,喷铂,置于SUPRA 55 Sapphire型场发射扫描电子显微镜下观测样品形貌,电镜放大倍率主要为5 K,20 K和50 K倍。
1.2.3.2 傅里叶红外变换光谱分析(FTIR)
采用溴化钾压片法取0.001 g干燥的样品,与0.05 g干燥的KBr粉末混合,充分研磨后,转移混合粉末于压模槽中,在压片机中调节液压压力为7 KPa,将混合粉末压制成薄片,将压片放入MAGNA-IR550型傅立叶变换红外光谱仪,于4000~500 cm-1波长范围内进行扫描测试。分析样品对红外吸收特性。
1.2.3.3 X-射线衍射测定(XRD)
使用Rigaku miniflex 600型 X射线衍射仪测定,操作电压为40 kV、电流强度为10mV,2θ扫描范围为3~80°。用于分析样品的微观结构。
1.2.3.4 比表面积(BET)
使用ASAP2460型多站全自动比表面积及孔径测试系统,以N2为吸附气体,进行全孔检测,脱气温度为250℃,脱气时间为7 h。对样品的比表面积大小和孔径分布进行检测。
2 结果与讨论
2.1 淀粉碳化物/海泡石复合材料制备工艺的单因素优化
2.1.1 淀粉与海泡石质量比对复合材料吸附量的影响
由图1可得,在质量比为1.5:1.0~2.5:1.0范围内,对亚甲基蓝的吸附量呈现增高的趋势,这是由于适当提高原料投加,固相产物的碳元素含量提高,海泡石上负载的碳化微球量相对增加,从而提高吸附量。而当质量比提高到2.5:1.0~3.5:1.0范围内时,吸附量呈现降低趋势,且下降程度趋于平缓,这是由于反应釜中存在的过量淀粉在被碳化之前会大量团聚,导致团聚体内部无法碳化完全,会使固相碳含量减少的同时,会占据大量空间,阻碍淀粉碳化物与海泡石结合作用;另外,淀粉量添加过多,导致反应釜中投料量过多,反应釜内部压力改变减少了碳化微球的活性基团的产生,影响复合材料的吸附效果。因此当淀粉和海泡石质量比例为2.5:1时较佳,此时吸附值为39.50 mg/g。
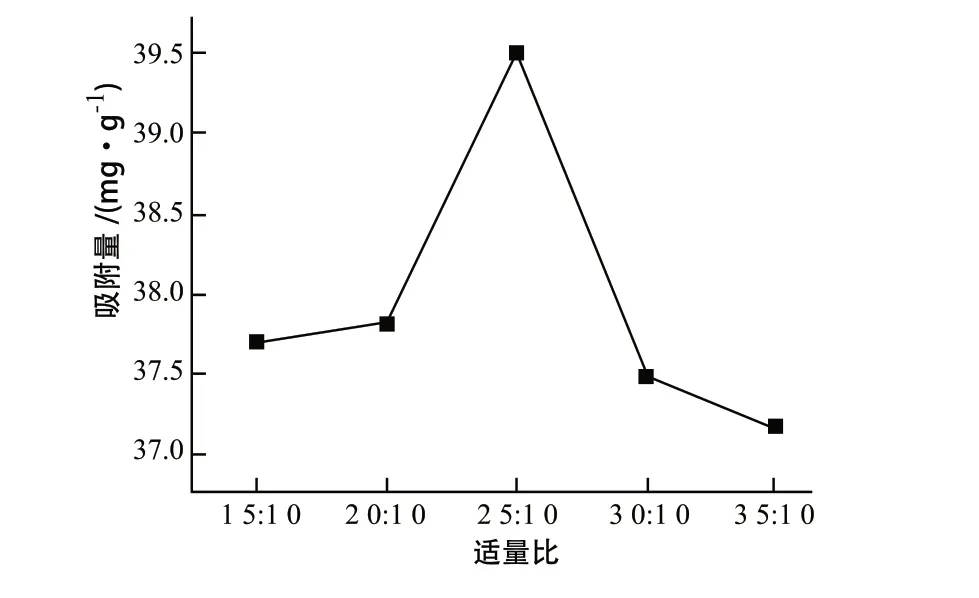
图1 淀粉与海泡石质量比对复合材料吸附量的影响曲线Fig.1 Effect of mass ratio of starch to sepiolite on adsorption capacity on composite materials
2.1.2 碳化时间对复合材料吸附量的影响
由图2可见,材料碳化时间为16 h时,所得样品在本批实验样品中吸附值达到最大,当反应时间少于16 h时,碳化反应未开始或者样品未碳化完全,无法生成或生成的活性功能团的数量少,导致所制成的复合材料吸附量减少。随着反应时间的延长(即反应时间超过16 h),淀粉碳化物表面的活性官能团的数量会随着减少[21];碳化反应所生成的焦油等多种副产物没有及时洗脱,在高温条件下,更容易被海泡石所吸附,堵塞海泡石孔隙而难以洗脱,导致复合材料吸附性能下降。故将16 h定为较佳碳化时间,此时样品吸附值为40.76 mg/g。

图2 碳化时间对复合材料吸附量的影响Fig. 2 Effect of carbonization time on the adsorption capacity on composite materials
2.1.3 碳化温度对复合材料吸附量的影响
由图3发现,在180~ 200℃的温度范围内,吸附量增高,这是因为在过低的温度条件下,淀粉热解脱水受到影响,导致其碳化程度不完全,影响了活性基团的产生,导致吸附量减低;并且随着体系温度升高,海泡石内部残留的结晶水逸出,复合材料孔隙更为通畅,吸附容量逐渐提高。而当碳化温度从200℃升高到220℃时,样品的吸附量逐渐减低,这是由于温度过高会导致副反应发生的同时,部分淀粉被降解完全,导致固相碳量减少,从而使复合材料的吸附性能呈现减弱的趋势。因此,200℃为反应的最优碳化温度,其吸附值为40.88 mg/g。

图3 碳化温度对复合材料吸附量的影响趋势曲线Fig. 3 Effect of carbonization temperature on the adsorption capacity of composite materials
2.2 响应面实验数据分析
根据单因素实验所得的数据作为响应面实验各项条件设置的基础。以复合材料对亚甲基蓝的吸附量(Y)为响应值,设定淀粉和海泡石质量比为因素A,碳化时间为因素B,碳化温度为因素C。最终,根据Box-Benhnken的中心组合实验设计原理设计出了(3因素3水平)响应面实验方案,组成响应面实验因素水平设计表见表1,最终响应面实验设计及相对应响应值表见表2[25-26],利用Design-Expert软件分析处理响应面实验所得到的数据,即得到响应面实验数据方差分析结果见表3。
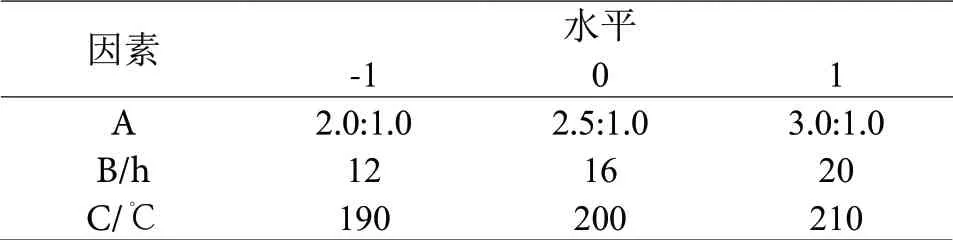
表1 响应面实验因素水平设计Table 1 Design of factors and levels of response surface experiments
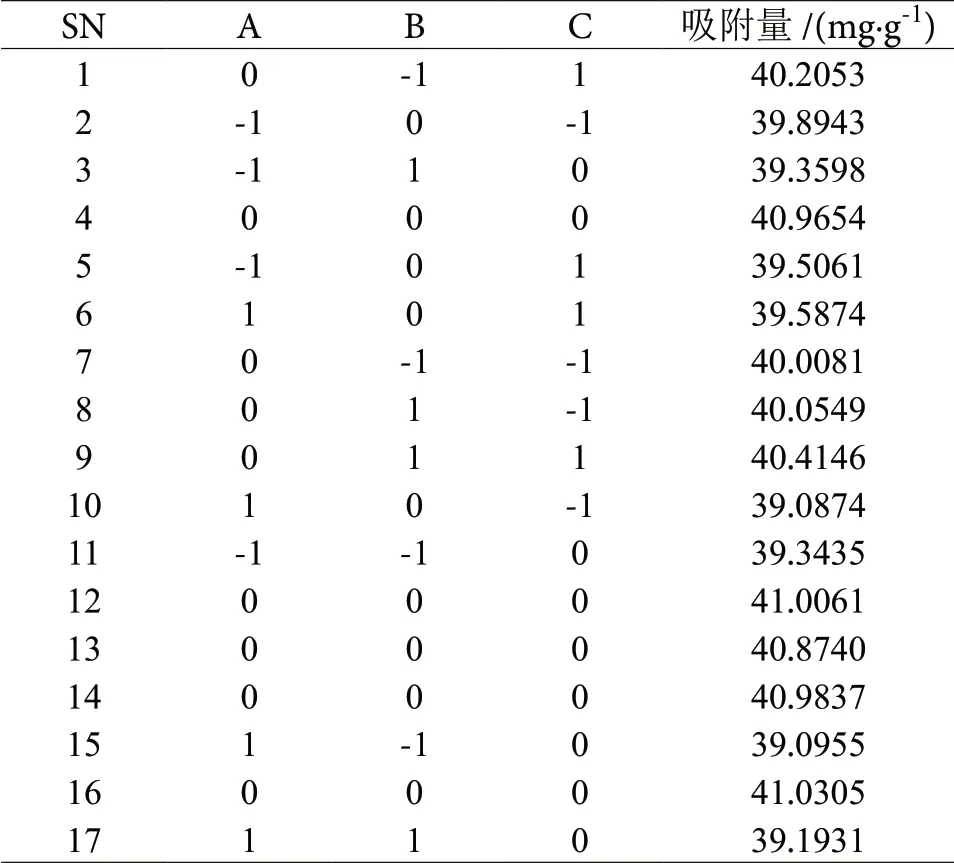
表2 响应面实验设计及相对应响应值Table 2 Response surface experimental design and corresponding response values

表3 响应面实验数据方差分析Table 3 Analysis of data variance of response surface experiments
P值和F值可以判断在实验中各变量对响应值(复合材料对亚甲基蓝的吸附量)影响的显著性。通过F检测进行影响显著性判断,在P值<0.01的情况下,判定变量对响应值的影响极显著,若P值<0.05,则认为该变量对响应值的影响显著,当P值>0.05,则变量对响应值影响不显著。
分析响应面实验数据可得回归的响应面二次多项式,见式(3)。
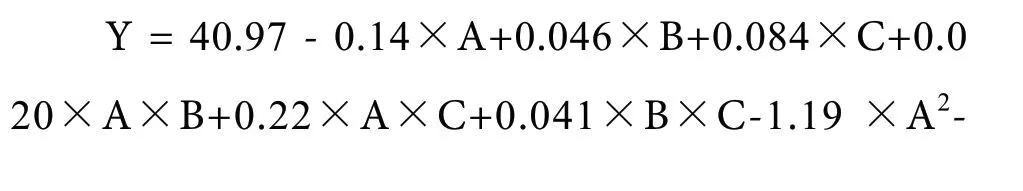

由表3发现,数学模型中“Model(模型)”项P<0.0001,为极显著;“Lack of Fit(失拟项)”为0.1387>0.05,即可说明该模型对解释各因素影响所导致响应值的变化的拟合优度高,数据处理误差小。“C”项对响应值的变化表现为显著,“A”、“AC”、“A2”、“B2”和“C2”各项均对响应面表现为极显著。响应面的二次多项式的相关系数为R2=0.9937,调整系数为R2Adj=0.9855,证明该模型与回归的响应面二次多项式的拟合程度高,可用于响应面分析计算。根据图4发现,各项实验数据基本上在中心线较小的范围内波动。因此,模型预测值与实验实测值较为相近,进一步说明,该响应面实验模型的拟合程度较高,可以应用于本次St-Sep复合材料的工艺条件的优化实验响应值预测。
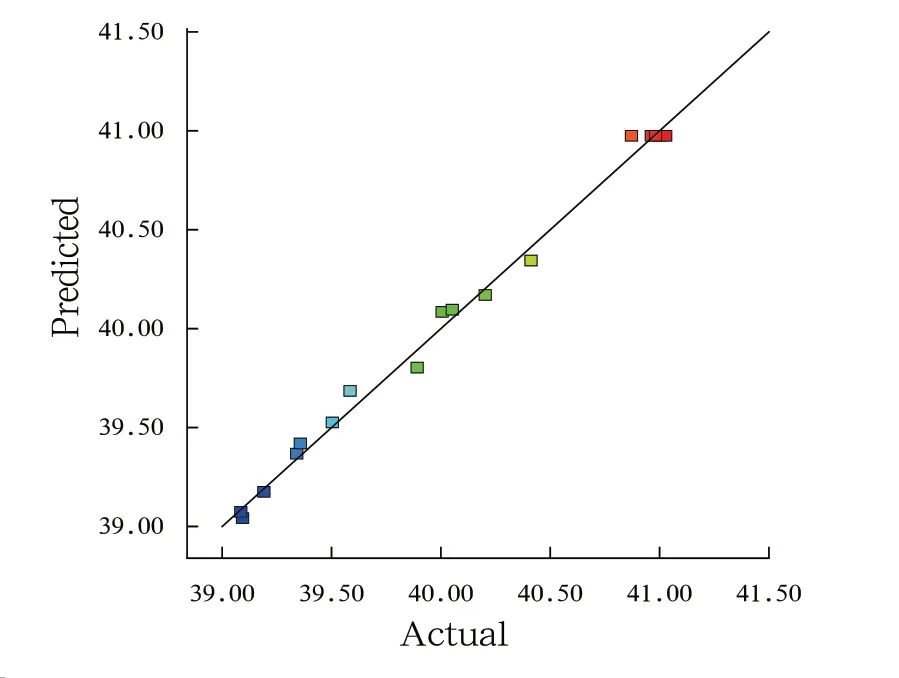
图4 复合材料对亚甲基蓝的吸附量实测值与预测值拟合Fig. 4 Fitting of actual and predicted values for the adsorption of MB by composites
由图5~7,各因素交互作用的3D曲面弧度和等值线的纵深跨度,结合方差分析F值,发现各因素对响应值的影响程度为:淀粉与海泡石质量比>碳化温度>碳化时间,且AC的交互作用最为显著,并且各因素对响应值的影响不是简单的线性关系。在实验范围内,各因素对响应值的影响呈现大致相同的变化趋势,即随着因素水平的提高,响应值先向高水平上移之后再下降的趋势,这与单因素实验结果相似。

图7 碳化时间与碳化温度(BC)间交互作用的3D响应曲面图与等值线Fig. 73D response surface plot and contour plot of the interaction between carbonization time and carbonization temperature (BC)
由响应面实验结果可确定St-Sep复合材料工艺最优工艺条件为:淀粉与海泡石质量比为2.5:1.0;碳化时间为16 h;碳化温度为200℃,预测较优吸附量为40.9719 mg/g。
在该最优条件下进行验证实验,其结果见表4。

表4 验证实验结果Table 4 Results of verification experiments
实测吸附量稍低于预测值0.89%,可以认为该数学模型能够较为准确地预测St-Sep复合材料对亚甲基蓝吸附量的预测,可用于St-Sep复合材料的制备工艺优化实验。
2.3 St-Sep复合材料性质表征
2.3.1 电镜分析结果(SEM)
图8为天然海泡石(A)、淀粉碳化物(B)、St-Sep复合材料(C)和St-Sep复合材料(D)的SEM图。

图8 三种材料的SEMFig.8 SEM images of three materials
由图8可以看出,在50 K倍电镜下,天然海泡石呈现为疏松多孔的纤维束状形貌(A),表面较为光滑且空隙较大。在5K倍电镜下,淀粉碳化物微球大致呈现球状(部分因粘结呈现类球状或葫芦状),直径约为2~4 μm,并且存在着粘连现象。在5 K倍电镜下游离的碳化微球的形貌和大小都与淀粉碳化物(B)相似,而在高倍率(50 K倍),原本光滑的海泡石表面变得粗糙,这是由于淀粉碳化物存在于海泡石纤维的表面上,在海泡石表面形成较为细密的碳化物层,并且碳化微球呈现非均匀分布,直径普遍变小,主要是由于:(1)海泡石独特的空间结构和作用力导致碳化微球总体变小;(2)水溶液中的淀粉分子或者淀粉在水热条件下分解而成的小分子酸[27],先被吸附到海泡石空隙中,再继续发生碳化反应。
由SEM图能够较为直观的发现,球型或类球型的淀粉碳化物成功的粘附于海泡石纤维的表面上,并且由于海泡石表面的碳化物层的存在,为St-Sep复合材料提供了更多的吸附作用位点。
2.3.2 红外光谱结果分析(FTIR)
图9为海泡石(a)、St-Sep复合材料(b)和淀粉碳化物(c)的红外光谱图。
由图9可看出,复合材料的红外特征吸收峰分布与原料海泡石相似。在3683~3010 cm-1之间出现较宽的吸收带,这是海泡石内部与镁离子相连的O-H,以及海泡石内水分子的多种O-H伸缩振动产生的吸收带,体现了其内部所存在的结晶水和吸附水。而1208 cm-1处则为O-H的弯曲振动峰。在1075~977 cm-1处表现出的较强的吸收带是Si-O伸缩振动所导致的[28]。
而复合材料与海泡石原料的红外谱图的区别在于,复合材料的谱图在2979~2924 cm-1处和1450~1345 cm-1处分别出现了饱和烃C-H的吸收弯曲振动和伸缩振动的吸收。在1705 cm-1和1615 cm-1处分别对应出现C=O和C=C的吸收峰。
在碳化后的淀粉的红外吸收谱图中在2979~2924、1450~1345、1705和1615cm-1处均有特征吸收出表现,因此,复合材料总体的红外谱图以海泡石的谱图为基础骨架,并且淀粉碳化物的特征吸收峰也出现,这表明复合材料的成功合成。
2.3.3 X射线衍射分析(XRD)
图10是海泡石、St-Sep复合材料和淀粉碳化物的XRD谱图。
由图10可知,在经过水热碳化复合前后,海泡石和St-Sep复合材料晶型基本无改变,但是与St-Sep复合材料的XRD图谱的峰强度略比海泡石的有所降弱,淀粉碳化物的图谱在20~23°之间形成无定型碳的宽化衍射峰,而在复合材料谱图中无定形碳的宽化衍射峰示出现,这可能是无定形碳宽的衍射峰被海泡石的较强的衍射峰所覆盖。
2.3.4 比表面积(BET)测定结果
从图11可以看出,海泡石与St-Sep复合材料的N2吸附-脱附等温线皆属于含有滞后环的IV型吸脱附等温线。St-Sep复合材料的比表面积为71.5390 m2/g。用BJH法求得在脱附过程中的平均孔径为20.9868 nm,天然海泡石孔容为0.622332 cm3/g,负载碳化物后孔容减小至0.293433cm3/g。由图12的孔径分布可得海泡石与St-Sep复合材料的孔径大部分分布在10~40 nm之间,属于介孔类型。
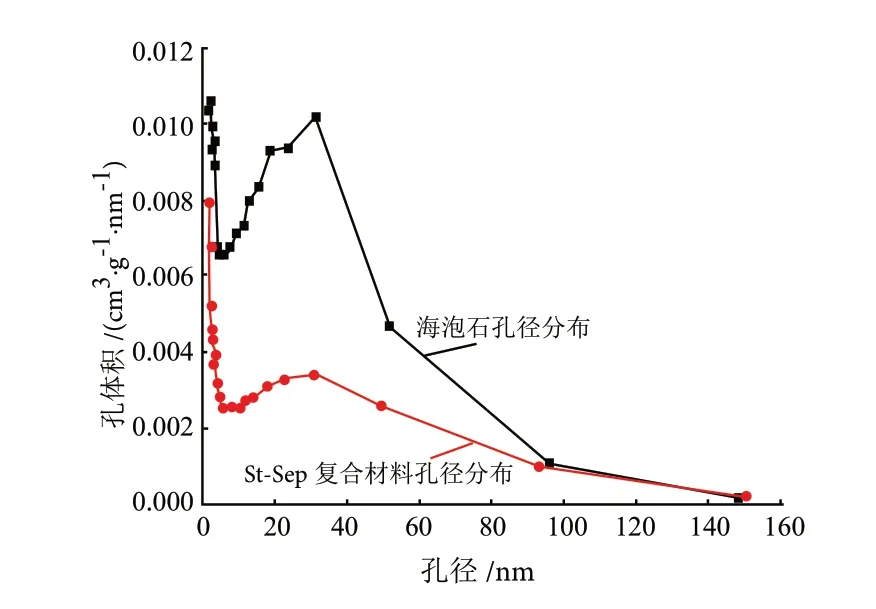
图12 海泡石与St-Sep海泡石复合材料的孔径分布情况Fig.12 Pore size distribution of sepiolite and St-Sep composites
由此可见,St-Sep复合材料的比表面积(71.5390 m2/g)明显小于海泡石的比表面积(181.5825 m2/g),这符合随着负载量的增而比表面积逐渐变小的趋势。这可解释为,淀粉碳化物在海泡石表面及空隙上形成细密的淀粉碳化物层后,堵塞了孔径较小的部分,同时导致复合材料总体孔容减小。这与SEM表征结果相符,也证明在材料复合后,海泡石纤维表面负载有淀粉碳化物。
3 结论
(1)扫描电镜分析结果表明,均匀且细密的碳化微球层分布在St-Sep复合材料表面,且碳化微球的直径普遍变小,直径约为0.10~0.15 μm,说明:海泡石独特的空间结构和作用力导致碳化微球总体变小;水溶液中的淀粉分子或者淀粉在水热条件下分解成小分子酸,先被吸附到海泡石空隙中,再继续发生的碳化反应。红外光谱图表明,复合材料有饱和羟C-H、C=O和C=C双键的存在,并且以海泡石的谱图为基础骨架,并且淀粉碳化物的特征吸收峰也出现这说明复合材料以海泡石为载体,碳化微球为负载物;XRD图谱表明材料复合前后,晶型不发生改变;BET分析结果,符合随着负载量的增加而比表面积逐渐变小的趋势。由此可知,淀粉碳化物成功负载于海泡石表面上;在负载了淀粉碳化物之后,复合材料的比表面积明显小于海泡石的比表面积,但复合材料对亚甲基蓝的吸附能力明显高于海泡石,说明了淀粉碳化物负载在海泡石表面后,所引入的C-H、C=O和C=C等有机活性官能团,增强了复合材料的吸附能力。
(2)响应面法确定St-Sep复合材料工艺最优工艺条件为:淀粉与海泡石质量比为2.5:1.0;碳化时间为16 h;碳化温度为200℃,最优吸附量为40.6084 mg/g,吸附效果优于经过水热碳化处理的海泡石原材料(吸附量为35.13mg/g)。单因素实验与响应面实验结果表明,各因素对亚甲基蓝的吸附量的影响大小依次为:淀粉与海泡石质量比>碳化温度>碳化时间。
(3)淀粉碳化物/海泡石复合材料的制备过程简便,且比用各种有机和无机试剂对海泡石改性的试剂用量少,能耗低,且造成二次污染的可能性更低。
