畜禽全基因组长纯合片段检测的研究进展
2022-01-17张鹏飞史良玉刘家鑫李洋吴成斌王立贤赵福平
张鹏飞,史良玉,刘家鑫,李洋,吴成斌,王立贤,赵福平
畜禽全基因组长纯合片段检测的研究进展
张鹏飞1,史良玉1,刘家鑫1,李洋1,吴成斌2,王立贤1,赵福平1
1中国农业科学院北京畜牧兽医研究所/农业部动物遗传育种与繁殖(家禽)重点实验室,北京 100193;2北京市昌平区动物卫生监督管理局马池口防疫站,北京 102202
长纯合片段(runs of homozygosity, ROH)是在个体和群体中常见的连续性纯合片段,是亲代将同源相同的单倍型遗传给同一个后代而形成的。ROH蕴藏着种群丰富的遗传信息,这使ROH成为一种有用的工具,可以提供关于种群是如何随着时间的演变而变化的信息。ROH也可以用于估计个体间遗传关系,有助于将近亲交配率降至最低,还可以暴露基因组中有害的变异。ROH在基因组中的大小、分布和频率受到自然选择和人工选择、重组、连锁不平衡、群体历史、突变率和近交水平等诸多因素的影响。近年来,随着高通量基因分型技术的使用以及二代测序成本的降低,畜禽育种已经进入基因组时代。对优秀种畜禽的选择强度大大提高,这在改善畜禽生产性能的同时不可避免地会造成动物的近交,从而导致近交衰退。根据ROH的分子信息能更准确地估计纯合性并可以检测过去和最近的近亲交配情况。基于ROH计算近交系数(F)反映的是个体的真实近交系数,即为实现的近交系数,而系谱近交系数PED得到的是期望值。F在缺乏系谱信息的情况下,也可以用来推断一个群体的历史和近亲交配水平的信息。选择会改变优良畜禽的表型,并重塑了基因组不同区域的ROH模式。此外,选择增加了目标位点周围的纯合性,有害的变异被认为更频繁地出现在ROH区域,可以通过ROH检测,降低复杂疾病发生的风险。经过长期选择,品种内同群个体相同ROH在基因组中高频出现,产生ROH岛。研究证实了ROH和正在选择的基因组区域之间具有相关性。在实际应用中,可以通过生物信息的方法在ROH岛注释到ROH区域与经济性状相关的基因。此外,ROH也为评估畜禽遗传多样性提供了新的视角,对群体进行全基因组ROH检测,剖析每个群体的遗传结构,并利用ROH对当前育种计划中近交的影响进行评估,来调整育种方案,保护品种的遗传多样性。ROH已逐渐成为探究群体历史结构、评估近交水平、鉴定候选基因方面的重要指标。识别ROH主要有观察基因型计数法和基于模型分析两种方法。常用的检测软件有PLINK、GERMLINE、BEAGLE、GARLIC等。在实际应用中,PLINK是最常用的ROH检测工具。在畜禽中,由于牛的SNP芯片推出最早,因此最先开展ROH研究的是牛的群体,牛的ROH研究数量最多、最深入。目前,在猪、羊、鸡等畜禽中关于ROH的研究也逐渐增多。文章主要综述了ROH形成的原理和检测方法,以及在畜禽中的研究进展,以期为畜禽的遗传育种提供参考。
长纯合片段;畜禽育种;单核苷酸多态性;近交评估;候选基因鉴定
0 引言
长纯合片段(runs of homozygosity, ROH)是二倍体生物中亲代将同源相同(identical by decent, IBD)的单倍型遗传给同一个后代,在该个体的基因组上形成的连续的纯合基因型区域[1],又称为染色体区段纯合子(chromosome segment homozygosity, CSH)[2]。1999年,Broman & Weber[3]首先在人类染色体上发现了ROH,并推断其可能影响人类健康。Gibson等[4]首次利用高密度SNP芯片在人类基因组中鉴定ROH,并研究了ROH在基因组中的分布情况等。此后,ROH才受到重视,激发了许多关于群体遗传学中ROH分析的研究。
因为ROH的长度、数量以及分布频率在不同个体基因组中并不相同,对这些独特的ROH进行分析,可以推断群体历史、评估遗传多样性、评估近交程度、促进畜禽遗传资源保护、鉴定候选基因和有害突变等。ROH已经逐渐成为量化和分析畜禽基因组近交水平、探究群体历史等方面重要指标。因此,本研究针对ROH形成的原理、影响因素、检测方法和在各畜禽品种中的研究进展进行综述[5]。
1 ROH产生的原理
在二倍体生物中,如果父母双方具有共同的祖先,并将从共同祖先中继承的相同染色体片段传递给同一后代,那么该个体在该片段内的所有基因型都是纯合的,从而产生ROH[6]。因此,ROH一般都是IBD基因组区段。ROH形成的过程如图1所示。从图1中可以看出,随着世代的增加,同源染色体之间不断发生重组,标记间的连锁不平衡(linkage disequilibrium, LD)不断被打断,造成ROH的长度也不断缩减。通过这个原理可以得知长ROH来源于亲缘关系较近的共同祖先,短ROH来源于较远的共同祖先,由此,以ROH长短来推断出个体之间的亲缘关系的远近。
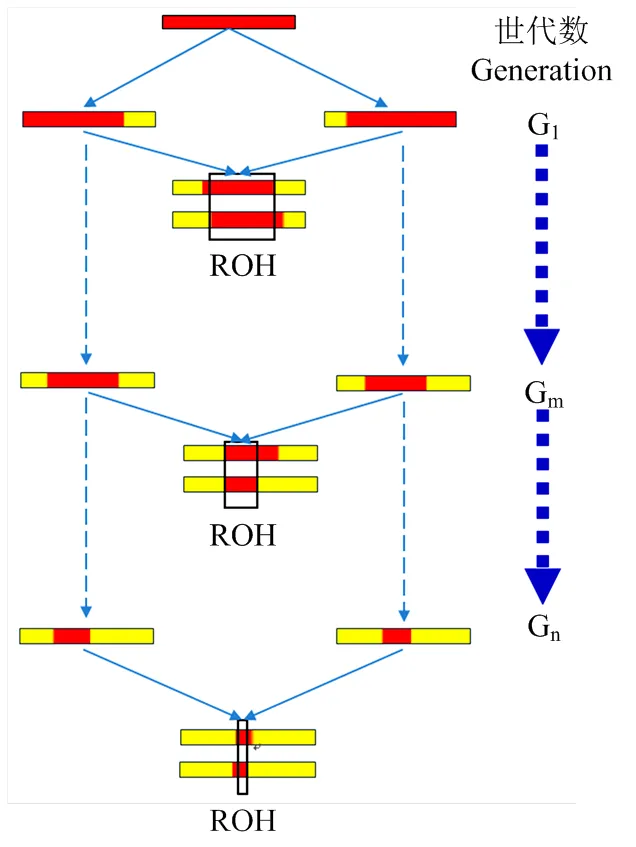
图1 ROH产生的原理
2 ROH的影响因素
从ROH形成的原理可知,影响ROH形成的因素很多,主要包括近交、选择、群体历史、漂变等。
2.1 近交
近交是ROH产生的最主要因素[7]。著名的进化论学家达尔文与表姐婚配之后,出生的子女早期的成活率明显低于正常人群,主要原因就是近亲导致子女继承了外祖父和外祖母长的IBD染色体片段,即ROH片段[8, 9]。意大利褐牛和荷斯坦牛比肉牛和兼用牛有更多和更长的ROH片段,表明其在更近世代发生过近交[10]。1974年,由新疆塔城地区的一头母牛和其子女,经过45年近交繁育,形成的新疆近交牛,其SNP位点纯合率高,导致ROH检出率提高,ROH 数量虽然较少,但是长ROH数量较多,ROH覆盖率(ROH%)也显著高于哈萨克等非近交牛群体[11]。
2.2 选择
选择会改变群体内等位基因的频率,包括基因组中ROH 的分布模式。在经过选择和未经过选择的北美荷斯坦奶牛中,通过ROH检测发现,经过选择的群体不仅ROH数目增加,而且ROH在基因组上的分布也不均匀,但是在非选择群体中,ROH在染色体上的分布变化不大[12]。
基因组选择技术加快了ROH的进行和群体的近交水平。北美荷斯坦奶牛在开展基因组选择后,Forutan等[13]比较了经过基因组选择和BLUP选择的动物个体内ROH的分布。与BLUP相比,基因组选择导致短ROH和中长ROH频率更高。基因组选择后ROH年增长率为2.1±0.05,而基因组选择前仅为0.57±0.01。这反映了与常规选择相比,基因组选择更能影响ROH的数量和分布。这也进一步说明在有限时间内基因组选择比常规BLUP选择更容易造成近交和产生ROH。
2.3 漂变
漂变会导致群体等位基因频率产生变化。在经历群体瓶颈后的种群,种群中存在的祖先谱系的数量会减少,ROH的数量会增加,但是ROH长度通常相对较短。相反,没有经历种群瓶颈的种群包含更多的祖先谱系,其ROH数量相对较少。
波兰长白猪和当地未选择的白猪和斑点猪在其历史上经历了严重的种群瓶颈,尤其是斑点猪。与波兰长白猪相比,白猪和斑点猪基因组中有着数量更多、更长的ROH,这一结果在所有ROH类别中都是一致的。而白猪和斑点猪的ROH近交系数也较高,在斑点猪中尤其高[14]。
2.4 群体历史
在品种形成过程中,受到群体结构、有效群体大小、交配方式等不同会导致动物体内形成不同的ROH分布模式。群体瓶颈增加了ROH数量,但这些ROH通常较短。一些种群既有瓶颈,又实行近交,因此有许多短ROH和长ROH,导致ROH分布最多[15]。奇灵厄姆牛就是极端纯合群体的一个例子。奇灵厄姆牛在过去的350年里没有经过选择,也没有经历过迁徙一直处于封闭状态。在其基因组中97.4%的SNP是纯合的,ROH基因组覆盖率为95%,14个ROH大于30Mb。表明该群体的遗传变异减少并具有极端纯合性[16]。
3 ROH的应用
3.1 评估近交
2008年,McQuillan等[17]提出将个体基因组所有ROH总长度占常染色体总长度的比值作为个体近交系数(F)。其计算公式为:,其中,
L是每条ROH的长度,L是常染色体总长度。
除此之外,还有三种方法计算近交系数。
第一种基于系谱的近交系数,该方法由Wright[18](1922)提出。计算公式为:

其中,F是个体的近交系数,N是个体的两亲本到共同祖先相关通径链的个数,F是共同祖先A的近交系数。
第二种,基于单个SNP计算的近交系数有两种计算方法。其一为检测基因型中纯合SNP所占比例(F1),其公式为[19]:

其中,N1为纯合SNP的个数,为检测SNP的总个数。其二计算方法为F2=(O(HOM)-E(HOM))/(1- E(HOM))[20],其中,为观测纯合子数目,为期望纯合子的数目,为SNP数目。
第三种,基于基因组关系矩阵计算近交系数[21]

其中,是SNP的数量,p是等位基因频率,x是第个SNP的拷贝数。
以上4种近交系数的计算方法,只有基于系谱的近交系数是依赖于系谱记录准确性,但是在实际中,系谱记录往往存在错误或缺失,而且在基础群中假设每个个体相互之间不存在亲缘关系。另外3种都是基于基因组的信息,其中F和F两种方法能得到-1—0的值,甚至会出现小于-1的值,F虽然控制在0—1之间,但是不能区分纯合等位基因是同源相同(identical by blood, IBD)还是同态相同(identical by state, IBS),得到的值会比真实值偏大。只有F是利用IBD来计算个体的真实近交系数,避免以上的问题,是目前研究中最有效的方法[22]。F和F之间存在强正相关,相关系数可达0.75[23],而且F还可以对特定染色体区段或者特定基因组区段进行近交系数估计[24]。F可能是评估动物亲缘关系和近交水平的一种有效且准确的替代方法[23]。
为了更好地理解F和F之间的区别,以自交的情况进行说明(图2),一个个体具有两条同源染色体,配子结合后产生2种类型的合子,其中F分别为1和0。按照系谱计算,自交群内所有个体的近交系数都为0.5,也可以通过群体每个个体的F值求期望而得到,即:1×1/2+0×1/2=0.5。从计算过程可以看出,F可以计算个体的真实近交系数,即为实现的近交系数,而系谱得到的是期望值。

图2 自交情况下两种近交系数估计值
3.2 推断群体历史
ROH在不同种群中会有不同分布。远交种群的ROH分布模式与其有效种群大小有关,小种群有着较多的ROH,较大的种群有着较少的ROH。而混合种群由于在两个或更多的祖先种群中有更远的共同祖先,所以比各自亲本种群具有更少的ROH。经历了种群瓶颈的群体具有更多的较短的ROH,反映了更深层次的亲缘关系[1]。通过ROH检测,可以从动物个体的基因组中ROH分布模式获得遗传背景信息,推测其种群形成历史,对研究其种群进化、品种形成具有重要意义[25]。
3.3 鉴定重要基因
畜禽品种经过长期的选择而形成,使得品种内同群个体间继承了一个共同祖先相同的染色体片段增加,使得相同ROH在多个个体基因组中出现高频的ROH,其在所选择的区域周围具有高水平的纯合性,称为ROH岛(图3)。ROH岛就是群体内ROH富集的区段,ROH岛不是随机分布在整个基因组上,而是在一个品种内的个体之间共享,研究证实了ROH和正在选择的基因组区域之间具有相关性[26]。高频率ROH的基因组区域可以通过生物信息的方法注释到与经济性状相关的基因[27]。因此,可以通过ROH岛进行定位候选基因。
ROH区段也会增加隐性纯合子量,使得隐性疾病的发生。Garrod等[28]观察到近交个体中隐性疾病的高发病率是由于它们对于有害的隐性等位基因遗传的IBD是纯合子的可能性很高。有害的隐性变异可以通过研究ROH在近交系个体中识别出来。有害纯合子变异的频率与基因组ROH之间存在很强的线性关系(0.93—0.98)。ROH覆盖率高的个体在长ROH中出现有害变异的比例更高[29-30]。因此,可以通过鉴定ROH,减少复杂疾病发生的风险。
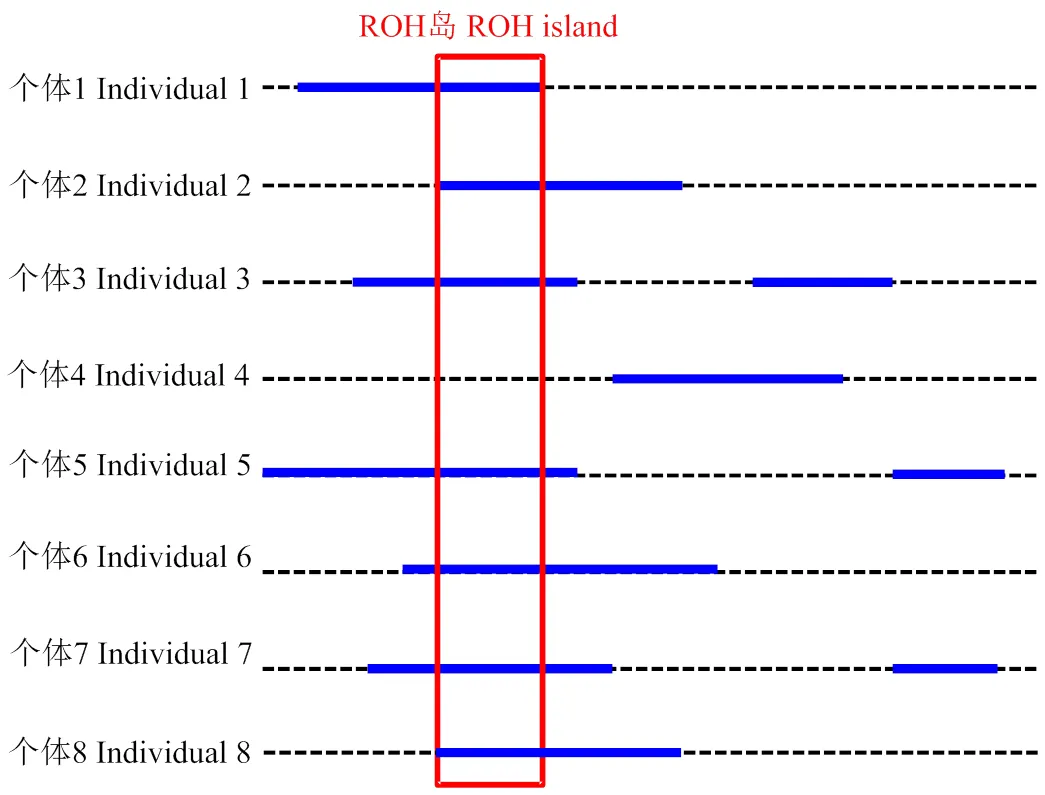
图3 ROH岛
3.4 评估遗传多样性
高通量基因分型阵列的出现极大地促进了遗传多样性研究[31-32],基因组ROH为评估畜禽遗传多样性提供新的思路方法。ROH区段使得群体纯合子增加,降低了遗传多样性,通过对畜禽进行全基因组ROH检测,剖析每个群体的遗传结构,并针对当前育种计划对近交的影响进行评估,来调整育种方案[33]。
3.5 基因组选择
基因组选择的基础是准确估计训练群和候选群之间的基因组关系,以获得准确的基因组估计育种值(GEBV)。Luan等[34]提出了利用ROH构建基因组关系矩阵(GROH)预测GEBV,并通过模拟比较了利用GROH、基于全群体连锁不平衡关系(GIBS)和基于连锁分析关系(GLA)预测的GEBV的准确性和偏差。结果表明,GROH估计GEBV始终比GIBS和GLA高0—40%。在真实数据中,GROH和GLA具有类似的准确性。
4 ROH检测方法
目前,识别ROH有两种主要方法:观察基因型计数法和基于模型分析。实际应用中,需要根据不同数据的特点,采用不同的算法。
4.1 观察基因型计数法
观察基因型计数法是指定每个窗口可变数量的杂合或缺失SNP,沿染色体长度方向移动固定大小的窗口来扫描每条染色体,以寻找在特定长度上显示纯合子的连续SNP。常用的检测软件有PLINK[35-36]、GERMLINE[37]。
4.2 基于模型分析
基于模型分析的方法主要利用隐马可夫模型来鉴定基因组中纯合子和杂合子区域,并获得重组率等基因组参数。常用的软件主要有BEAGLE[38]、GARLIC[39]。
4.3 两种检测方法效果的比较
在实际应用中,观察基因型计数法应用较为广泛,相关研究也较多。PLINK是最常用的软件。Howrigan[40]等使用PLINK、GERMLINE和BEAGLE进行了ROH分析。结果发现PLINK的滑动窗口算法在使用针对连锁不平衡(LD)进行修剪的SNP数据时效果最好。但是观察基因型计数仅使用基因型数据,如果不进一步对样本中的缺失和重复进行分析,则不能区分由单拷贝缺失引起的真实纯合性和虚假纯合性[41]。
基于模型分析方法更适合序列数据,可以从群体测序数据获得关于等位基因频率和重组率的更全面的信息,具有更高的灵敏度[42]。随着测序技术的广泛应用和成本下降,基于模型分析方法会提高ROH识别准确性,降低错误率。
5 ROH在畜禽中应用的研究进展
ROH已经广泛应用于畜禽品种的种群研究中。由于牛的SNP芯片推出最早,因此最先开展ROH研究的是牛的群体。目前,在猪、羊、鸡等畜禽中关于ROH的研究也逐渐增多,以下将分别详细介绍。
5.1 ROH在牛中应用的研究进展
5.1.1 评估近交 Ferencakovic等[43]首先在牛上进行了关于ROH的研究,分析了554头奥地利弗莱克维公牛精液中近交抑制的遗传特征与活精子百分率和精子总数的可能关系。分别计算了系谱近交系数和4种不同片段大小的ROH近交系数,其中F检测到精子总数存在显著的近交抑制(<0.05)。在之前研究中,基于ΔAIC,F是最好的评估精子总数的近交衰退的指标[44],而F的ΔAIC值为3.9,表明F可用于近交衰退的检测。杨湛澄等[19]利用牛54 K SNP 芯片数据计算了2 107头荷斯坦牛的F、F和F。F与F、F之间的相关系数分别为0.46和0.38。F与F秩相关和Pearson相关分别为0.97、0.95,表明F与F之间高度相关。F与F、F所有秩相关均较高(0.72— 0.97),且>0.01。研究表明在缺失系谱数据的情况下,基因组近交系数是评估个体近交水平更为准确的工具,也为以后研究奶牛近交衰退和与疾病关联ROH区域奠定基础。Forutan等[13]计算了北美荷斯坦牛的ROH近交系数,并比较了不同选择方法对ROH模式的影响。当SNP窗口设置为20—50个SNP时,在所有情况和所有选择标准下,F比F和F更接近真实近交系数。师睿等[11]对新疆近交牛群计算了F和F两种基因组近交系数来验证新疆近交牛在基因组上高度纯合,基于ROH覆盖率计算的近交系数为0.136,明显高于其他种群(0.021—0.102)。Xu等[45]用F、F和F3种方法估计了不同中国地方黄牛品种的每个群体内的近交系数。个体F在0.01—0.34之间变化,F、F和F之间存在较高的相关性(0.78—0.85),F与F的相关性最高(= 0.85,<2.2×10−16),F与F(=0.78,<2.2×10−16)呈显著正相关。F与F和F之间的高度相关性可以被认为是IBD基因组比例的准确估计值。
5.1.2 推断群体历史 Purfield等[23]分析了891头多个品种牛的ROH模式,观察到ROH在不同品种之间存在显著差异。荷斯坦、荷斯坦-弗里西亚和弗里西亚品种在较长的ROH中显示出最大的覆盖率,平均为700.3Mb。这反映了最近世代的近交。研究表明ROH分析量化了基因组变异特征,可用于推断种群历史。Ferencakovic等[46]研究了几个不同品种牛的纯合性,发现ROH的分布和频率在不同品种之间存在差异。瑞士棕色牛的ROH平均数最高(98.9),ROH的基因组覆盖率最高。弗莱克维公牛的平均ROH长度最短,主要由许多短ROH组成。瑞士棕色牛长ROH较少。挪威红牛与弗莱克维公牛有着相似的分布模式。通过ROH的不同分布,推断出瑞士棕色牛中观察到的大量长ROH与从少量公牛进口精液有关。弗莱克维公牛显示出很小的纯合子比例的基因组,这与较大的有效种群规模一致。在挪威红牛中发现的ROH分布模式被归因于该品种历史上的混合所导致的高度异质性。中国地方牛的ROH模式及其分布在很大程度上仍未被探索。Xu等[45]在南大牛和文山牛中发现了数量较多的长ROH(100—250 Mb),这种ROH模式可能是由于南大牛和文山牛的种群相对较小,经历了瓶颈。延黄牛有着较少的ROH和短ROH,这与最近在延黄牛的个体中混杂的情况是一致的。柴达木牛和平武牛的ROH的总长度和数目都处于中等水平。这可能反映了这些品种中没有混杂。研究利用ROH推断群体历史有助于揭示黄牛品种间的遗传多样性[45]。
5.1.3 鉴定重要基因 Ferencakovic等[43]分析了奥地利弗莱维赫公牛导致近交衰退的基因组区域,发现精子总数和活精子百分比分别与4个ROH区域和5个区域显著相关。除一个区域外,所有区域都鉴定到与精子发生和精子形态相关的基因。如与严重的生精衰竭有关的、影响精子活力的和在精子渗透调节中发挥重要作用的。Mastrangelo等[47]对4个奶牛品种的ROH岛进行候选基因鉴定。共鉴定出126—347个基因,如与生殖性状有关的和与乳腺功能有关的,以及与免疫系统有关的。其中,在一个ROH岛上定位到一个QTL,包含6个基因(),并注释到与产奶性状相关的、。Xu等[45]在中国地方黄牛品种中的ROH岛中鉴定到了一些与免疫相关的。在柴达木牛中的ROH岛中鉴定到了与脂肪酸有关——可能调节肉类嫩度的。在延黄牛中检测到与能量肉质、动态平衡和多糖有关的和等。在凉山牛品种中检测到与肉牛的肌脂组成和大理石纹有关的和。此外,在平武牛、文山牛和昭通牛中分别鉴定了与生长性状和免疫过程相关的品种特异性基因和。
5.2 ROH在猪中应用的研究进展
5.2.1 评估近交 Gomez-Raya等[48]首次提出了将ROH长度作为随机变量的新型近交系数。使用伊比利亚猪品系的217头母猪比较了新型近交系数和系谱近交系数。两种近交系数的相关系数在0.60—0.70,此外,研究发现近交系数受染色体长度的影响很大,二者存在显著相关。利用ROH长度分布来测量近亲交配,不仅可以识别染色体近交,而且还可以提供有关最近世代近交的信息,为更准确地评估近交对伊比利亚猪封闭品系的影响奠定了基础。Shi等[49]研究了3 692头大白猪ROH的发生和分布,计算并比较了系谱近交系数F、基因型中纯合SNP所占比例F、基因组近交系数F和4种类型的F(1—5Mb、5—10Mb、大于10Mb、总ROH)。F与F的相关系数最高(0.95),而F与F的相关系数最低(0.083)。F与F的相关系数在0.18—0.37之间,除>10Mb的ROH外,F与F的相关性随ROH长度的增加而增大。与其他近交系数相比,F可准确评估个体近交水平。在没有系谱的情况下,F是近交估计的另一种选择。
5.2.2 推断群体历史 Bosse等[50]对欧亚大陆野猪和商品猪的全基因组的ROH进行检测。发现来自不同群体的个体基因组中ROH长度、数量和分布模式上存在明显差异。但同一种群的动物基因组中有着相似的ROH模式。在驯化的亚洲猪中,长ROH数量最多,这可能表明最近种群数量减少。欧洲野猪基因组中ROH数目最多,全基因组纯合子比例最高,这与欧洲冰川造成的群体瓶颈比亚洲更严重的证据是一致的[51]。Herrero-Medrano等[52]对伊比利亚半岛的野猪和驯养猪进行了全基因组ROH检测。野生种群和驯化种群在ROH分布模式有着显著差异。家猪Chato Murciano具有最多的长ROH数量,表明该品种最近的近交和低遗传多样性。野猪有非常多的短ROH、没有长ROH,这可能与过去种群规模的减少和最近的近交很少有关。这种ROH模式可能源于上个世纪欧洲的人口瓶颈,导致有效群体数量急剧减少。其他可能性包括亚种群的形成和动物的迁徙,与家猪的随机杂交,以及伊比利亚半岛没有地理障碍,阻止了野猪的高度近交[53]。
5.2.3 鉴定重要基因 Zhang等[54]对中国和西方猪的ROH岛进行检测和注释。在中国猪的ROH岛上鉴定到了与繁殖、免疫、肉质和适应性有关的基因,以及与西方猪的生长速度和免疫力有关的相关基因。如:与仔猪出生和死亡总数的和鉴定到与生长和脊椎数相关的。Xie等[55]对长白猪、松辽黑猪和约克夏猪3个群体ROH出现频率较高的基因组区域进行基因注释。鉴定出与繁殖性状()、肉质性状()和能量转换()相关的基因位于ROH高发区。Shi等[49]在大白猪的基因组中鉴定出了12个ROH岛,鉴定出许多控制大白猪重要经济性状的候选基因,其中一些已被证明是肌肉发育和脂肪沉积的重要候选基因,包括抑制精子与卵母细胞的结合的、参与调节猪的生殖行为的等。Szmatoła等[14]对波兰长白猪和未选择的白猪和斑点猪ROH岛进行基因鉴定。在波兰长白猪中,检测到与细胞葡萄糖稳态相关的生物学过程()和染色质沉默的调节()相关的基因显著丰富。在白猪中检测到168与细胞对生长因子刺激的反应的负面调控有关的基因()。
5.3 ROH在羊中应用的研究进展.
5.3.1 评估近交 Purfield等[56]计算了6个商品绵羊的系谱近交系数、ROH近交系数、标记基因型中纯合子所占比例、基因组近交系数。Vendeen种群的F和F的相关性最低(0.12—0.15),Belclare种群的相关性最强(0.71—0.76)。F与F、F与F之间的相关性高于F与F之间的相关性,所有ROH近交指标对F的回归截距均大于零,表明与F相比,用F估计基因组纯合性更准确。刘家鑫等[57]利用50K芯片对10个绵羊群体的440个个体进行全基因组ROH检测,通过计算ROH基因组近交系数F探究其近交水平。结果发现不同品种基因组中ROH的分布明显不同,其中,国外种群(杜泊羊和德美羊)的平均F显著高于地方品种。地方品种中,藏羊平均F最高(0.085),苏尼特羊平均F最低(0.010)。
5.3.2 推断群体历史 Mastrangelo等[58]首次研究了ROH在多个具有不同的近交背景、选择历史和育种目标的绵羊品种中的发生和分布,结果发现,不同种群基因组内ROH分布明显不同。在科米萨纳和贝尔加莫品种中发现长ROH数量较少,这与之前研究结果一致,这反映了这两个品种最近世代的近交以及有着较大的有效群体规模。与其他品种相比,这两个品种的有效群体规模更大(571、557)。另一方面,巴雷斯卡、莱切塞和伯利兹谷品种的ROH分布模式表明有效种群规模较小(183、512、340),这在先前的研究中证实。科米萨纳和巴雷斯卡是亲缘关系较差的品种,其中,巴雷斯卡、莱切塞和伯利兹谷绵羊长ROH片段较多,反映了莱切塞和伯利兹谷绵羊最近世代的近交。
5.3.3 鉴定重要基因 Mastrangelo等[59]鉴定了516只贝利斯谷绵羊ROH频率较高的基因组区域,筛选出107个与产奶量和免疫反应有关潜在候选基因。包括与绵羊朊病毒蛋白沉积相关的,调节新陈代谢、体温、血压、内分泌和免疫功能等功能的,在血管收缩和血压调节中发挥重要作用的。Mastrangelo等[58]在意大利绵羊品种的ROH岛鉴定到在骨骼肌和猪肉品质中发挥关键作用的,影响饲料转化、肌肉生长和肥胖性状的,与牛的生长、脂肪沉积和肉类生产相关的,参与控制脂肪代谢和胰岛素敏感性的。刘家鑫等[57]在德美羊、杜泊羊、大尾寒羊等国内外不同绵羊品种的基因组中高频ROH区域鉴定到了26个与动物经济性状相关的基因,包括与绵羊生长发育相关的,与绵羊断奶重相关的,与肉品质、繁殖性状相关的。
5.3.4 评估遗传多样性 在家畜物种中,人们致力于揭示主要商业品种之间的遗传差异、关系和种群结构,但对不太广泛使用的地方品种的研究通常还不够深入。Beynon等[60]使用单倍型纯合性(HHN)方法对18个威尔士地方绵羊品种的遗传结构进行了特征分析,推断威尔士绵羊品种的群体历史和结构。这种方法依赖于ROH的全基因组分布。使用HHN的推断确定了低有效种群规模和高水平近交的品种,通过有计划的育种策略,间接的为遗传多样性的监测和恢复提供了信息。
5.4 ROH在鸡中应用的研究进展
Fleming等[61]使用600K SNP芯片对非洲的3个鸡群体进行了全基因组ROH检测。ROH数量和大小在个体中差异很大。黑鸡ROH最少,而乌干达土鸡有着较长的ROH。对3个群体中150个相同的ROH区域用GO富集进行分析,鉴定到与内源性和外源性应激源相关的通路。ROH分析的结果表明,这些种群的基因组受到了来自环境的选择压力。Zhang等[62]对白耳黄鸡、北京油鸡和狼山鸡3个保种群体进行了全基因组ROH检测。计算了传统近交系数和ROH近交系数。ROH与ES呈显著正相关(2=0.76)。北京油鸡ROH最高,表明该品种最近发生了近交。这反映了该品种较小的有效种群数量。白耳黄鸡、狼山鸡近交水平相近且较低(—0.05)。研究利用ROH来识别近代或古代近交留下的痕迹,避免了近交,这为保护种群的遗传多样性动态提供了新的思路,并为改进保护方案奠定了基础。
6 结语
基于ROH计算个体近交系数,不仅摆脱了对系谱的依赖,还能计算出特定染色体区域的近交情况,为评估群体和特定染色体区段的近交衰退程度[62-65],以及鉴定与ROH相关联的重要经济性状[66]提供了可行性。ROH已被证明在分析和量化基因组近交水平方面非常重要。此外,ROH研究在定位与疾病发生相关的隐性等位基因、探究种群历史及其结构、鉴定与重要性状相关的候选基因、减少有害变异的发生等方面发挥重要作用。这不仅为实际的育种和保种设置合理的交配方案提供参考依据,也为揭示重要经济性状的遗传机制提供了新的研究思路。
然而,现在利用ROH构建的个体间亲缘关系矩阵开展基因组选择还未见有实际数据报道。下一代测序平台的出现,使全基因组测序得以更低的成本和更短的时间实现。相比于SNP芯片,高通量技术和全基因组测序技术相结合,鉴定ROH的参数设置会发生明显变化,这对利用高通量的测序数据进行精确和快速的畜禽ROH检测提出了新的挑战。
[1] FRANCISCO C. CEBALLOS P K J, JAMES F. WILSON, DAVID W. CLARK, MICHèLE RAMSAY. Runs of homozygosity: windows into population history and trait architecture. Nature Reviews Genetics, 2018, 19(4): 220-234.
[2] HAYES M G, KELLY A L. High pressure homogenisation of milk (b) effects on indigenous enzymatic activity. Journal of Dairy Research, 2003, 70(3): 307-313.
[3] BROMAN K W, WEBER J L. Long homozygous chromosomal segments in reference families from the centre d'Etude du polymorphisme humain. American Journal of Human Gentics, 1999, 65(6): 1493-1500.
[4] GIBSON J, MORTON N E, COLLINS A. Extended tracts of homozygosity in outbred human populations. Human Molecular Genetics, 2006, 15(5): 789-795.
[5] 刘刚, 孙飞舟, 朱芳贤, 冯海永, 韩旭. 连续性纯合片段在畜禽基因组研究中的应用. 遗传, 2019, 41(4): 304-317.
LIU G, SUN F Z, ZHU F X, FENG H Y, HAN X. Runs of homozygosity and its application on livestock genome study. Hereditas(Beijing), 2019, 41(4): 304-317. (in Chinese)
[6] KIRIN M, MCQUILLAN R, FRANKLIN C S, CAMPBELL H, MCKEIGUE P M, WILSON J F. Genomic runs of homozygosity record population history and consanguinity. PLoS One, 2010, 5(11): e13996.
[7] CHARLESWORTH D, WILLIS J H. The genetics of inbreeding depression. Nature Reviews Genetics, 2009, 10(11): 783-796.
[8] ÁLVAREZ G, CEBALLOS F, BERRA FLS T. Darwin was right: inbreeding depression on malefertility in the Darwin family. Biological Journal of the Linnean Society, 2015, 114(2): 474-483.
[9] BERRA T M, ALVAREZ G, CEBALLOS F C. Was the Darwin/ Wedgwood Dynasty Adversely Affected by Consanguinity? BioScience, 2010, 60(5): 376-383.
[10] MARRAS G, GASPA G, SORBOLINI S, DIMAURO C, AJMONE- MARSAN P, VALENTINI A, WILLIAMS J L, MACCIOTTA N P. Analysis of runs of homozygosity and their relationship with inbreeding in five cattle breeds farmed in Italy. Animal Genetics, 2015, 46(2): 110-121.
[11] 师睿, 张毅, 王雅春, 黄涛, 卢国昌, 岳涛, 卢振西, 黄锡霞, 卫新璞, 冯书堂, 陈军, 乌兰·卡格德尔, 茹先古丽·阿不力孜, 努尔胡马尔·木合塔尔. 利用SNP 芯片信息评估新疆近交牛基因组纯合度. 遗传, 2020, 42(5): 493-505.
SHI R, ZHANG Y, WANG Y C, HUANG T, LU G C, YUE T, LU Z X, HUANG X X, WEI X P, FENG S T, CHEN J, Wulan Kagedeer, Ruxianguli Abulizi, Nuerhumaer Muhetaer. The evaluation of genomic homozygosity for Xinjiang inbred population by SNP panels. Hereditas(Beijing), 2020, 42(5): 493-505. (in Chinese)
[12] KIM E S, COLE J B, HUSON H, WIGGANS G R, VAN TASSELL C P, CROOKER B A, LIU G, DA Y, SONSTEGARD T S. Effect of artificial selection on runs of homozygosity in u. s. Holstein cattle. PLoS One, 2013, 8(11): e80813.
[13] FORUTAN M, ANSARI MAHYARI S, BAES C, MELZER N, SCHENKEL F S, SARGOLZAEI M. Inbreeding and runs of homozygosity before and after genomic selection in North American Holstein Cattle. BMC Genomics, 2018, 19(1): 98.
[14] SZMATOLA T, JASIELCZUK I, SEMIK-GURGUL E, SZYNDLER- NEDZA M, BLICHARSKI T, SZULC K, SKRZYPCZAK E, GURGUL A. Detection of runs of homozygosity in conserved and commercial pig breeds in Poland. Journal of Animal Breeding and Genetics, 2020, 137(6): 571-580.
[15] CURIK I F, MAJA SöLKNER, JOHANN. Inbreeding and runs of homozygosity: A possible solution to an old problem. Livestock Science, 2014, 166(9): 26-34.
[16] WILLIAMS J L, HALL S J, DEL CORVO M, BALLINGALL K T, COLLI L, AJMONE MARSAN P, BISCARINI F. Inbreeding and purging at the genomic Level: the Chillingham cattle reveal extensive, non-random SNP heterozygosity. Animal Genetics, 2016, 47(1): 19-27.
[17] MCQUILLAN R, LEUTENEGGER A L, ABDEL-RAHMAN R, FRANKLIN C S, PERICIC M, BARAC-LAUC L, SMOLEJ- NARANCIC N, JANICIJEVIC B, POLASEK O, TENESA A, MACLEOD A K, FARRINGTON S M, RUDAN P, HAYWARD C, VITART V, RUDAN I, WILD S H, DUNLOP M G, WRIGHT A F, CAMPBELL H, WILSON J F. Runs of homozygosity in European populations. American Journal of Human Genetics, 2008, 83(3): 359-372.
[18] WRIGHT S. Coefficients of inbreeding and relationship. Amerlcan Naturalist, 1922, 56: 330-338.
[19] 杨湛澄, 黄河天, 闫青霞, 王雅春, 俞英, 陈绍祜, 孙东晓, 张胜利, 张毅. 利用高密度SNP标记分析中国荷斯坦牛基因组近交. 遗传, 2017, 39(1): 41-47.
YANG Z C, HUANG H T, YAN Q X, WANG Y C, YU Y, CHEN S H, SUN D X, ZHANG S L, ZHANG Y. Estimation of genomic inbreeding coefficients based on high-density snp markers in chinese holstein cattle. Hereditas(Beijing), 2017, 39(1): 41-47. (in chinese)
[20] YANG J, LEE S H, GODDARD M E, VISSCHER P M. GCTA: a tool for genome-wide complex trait analysis. American Journal of Human Genetics, 2011, 88(1): 76-82.
[21] VANRADEN P M. Efficient methods to compute genomic predictions. Journal of Dairy Science, 2008, 91(11): 4414-4423.
[22] BJELLAND D W, WEIGEL K A, VUKASINOVIC N, NKRUMAH J D. Evaluation of inbreeding depression in Holstein cattle using whole-genome SNP markers and alternative measures of genomic inbreeding. Journal of Dairy Science, 2013, 96(7): 4697-4706.
[23] PURFIELD D C, BERRY D P, MCPARLAND S, BRADLEY D G. Runs of homozygosity and population history in cattle. BMC Genetics, 2012, 14(13): 70.
[24] KELLER M C, VISSCHER P M, GODDARD M E. Quantification of inbreeding due to distant ancestors and its detection using dense single nucleotide polymorphism data. Genetics, 2011, 189(1): 237-249.
[25] PERIPOLLI E, MUNARI D P, SILVA M, LIMA A L F, IRGANG R, BALDI F. Runs of homozygosity: current knowledge and applications in livestock. Animal Genetics, 2017, 48(3): 255-271.
[26] PERIPOLLI E, STAFUZZA N B, MUNARI D P, LIMA A L F, IRGANG R, MACHADO M A, PANETTO J, VENTURA R V, BALDI F, DA SILVA M. Assessment of runs of homozygosity islands and estimates of genomic inbreeding in Gyr (Bos indicus) dairy cattle. BMC Genomics, 2018, 19(1): 34.
[27] LIU J X, SHI L Y, LI Y, CHEN L, DORIAN G, ZHAO F D. Estimates of genomic inbreeding and identification of candidate regions that differ between Chinese indigenous sheep breeds . Journal of Animal Science and Biotechnology, 2021, 12(1): 95.
[28] GARROD A E O M D L F R C P. The incidence of alkaptonuria: a study of chemical individuality . Molecular Medicine 1996, 2(3): 274-282.
[29] ZHANG Q, GULDBRANDTSEN B, BOSSE M, LUND M S, SAHANA G. Runs of homozygosity and distribution of functional variants in the cattle genome . BMC Genomics, 2015, 16(1): 542.
[30] SZPIECH Z A, XU J, PEMBERTON T J, PENG W, ZOLLNER S, ROSENBERG N A, LI J Z. Long runs of homozygosity are enriched for deleterious variation. American Journal of Human Genetics, 2013, 93(1): 90-102.
[31] KIM Y, RYU J, WOO J, KIM J B, KIM C Y, LEE C. Genome-wide association study reveals five nucleotide sequence variants for carcass traits in beef cattle . Animal Genetics, 2011, 42(4): 361-365.
[32] SNELLING W M, ALLAN M F, KEELE J W, KUEHN L A, MCDANELD T, SMITH T P, SONSTEGARD T S, THALLMAN R M, BENNETT G L. Genome-wide association study of growth in crossbred beef cattle . American Journal of Human Genetics, 2010, 88(3): 837-848.
[33] ISLAM R, LI Y, LIU X, BERIHULAY H, ABIED A, GEBRESELASSIE G, MA Q, MA Y. Genome-Wide Runs of Homozygosity, Effective Population Size, and Detection of Positive Selection Signatures in Six Chinese Goat Breeds . Genes, 2019, 10(11): 938.
[34] LUAN T, YU X, DOLEZAL M, BAGNATO A, MEUWISSEN T H. Genomic prediction based on runs of homozygosity . Genetics Selection Evolution, 2014, 46(1): 64.
[35] PURCELL S, NEALE B, TODD-BROWN K, THOMAS L, FERREIRA M A, BENDER D, MALLER J, SKLAR P, DE BAKKER P I, DALY M J, SHAM P C. PLINK: a tool set for whole-genome association and population-based linkage analyses. American Journal of Human Genetics, 2007, 81(3): 559-575.
[36] MEYERMANS R, GORSSEN W, BUYS N, JANSSENS S. How to study runs of homozygosity using PLINK? A guide for analyzing medium density SNP data in livestock and pet species . BMC Genomics, 2020, 21(1): 94.
[37] GUSEV A, LOWE J K, STOFFEL M, DALY M J, ALTSHULER D, BRESLOW J L, FRIEDMAN J M, PE'ER I. Whole population, genome-wide mapping of hidden relatedness . Genome Research, 2009, 19(2): 318-326.
[38] BROWNING B L, BROWNING S R. Detecting identity by descent and estimating genotype error rates in sequence data . American Journal of Human Genetics, 2013, 93(5): 840-851.
[39] SZPIECH Z A, BLANT A, PEMBERTON T J. GARLIC: Genomic Autozygosity Regions Likelihood-based Inference and Classification . Bioinformatics, 2017, 33(13): 2059-2062.
[40] HOWRIGAN D P, SIMONSON M A, KELLER M C. Detecting autozygosity through runs of homozygosity: a comparison of three autozygosity detection algorithms . BMC Genomics, 2011, 23(12): 460.
[41] KUNINGAS M, MCQUILLAN R, WILSON J F, HOFMAN A, VAN DUIJN C M, UITTERLINDEN A G, TIEMEIER H. Runs of homozygosity do not influence survival to old age . PLoS One, 2011, 6(7): e22580.
[42] NARASIMHAN V, DANECEK P, SCALLY A, XUE Y, TYLER-SMITH C, DURBIN R. BCFtools/RoH: a hidden Markov model approach for detecting autozygosity from next-generation sequencing data . Bioinformatics, 2016, 32(11): 1749-1751.
[43] FERENCAKOVIC M, SOLKNER J, KAPS M, CURIK I. Genome-wide mapping and estimation of inbreeding depression of semen quality traits in a cattle population . Journal of Dairy Science, 2017, 100(6): 4721-4730.
[44] MAXIMINI L, FUERST-WALTL B, GREDLER B, BAUMUNG R. Inbreeding depression on semen quality in Austrian dual-purpose simmental bulls . Reproduction in Domestic Animals, 2011, 46(1): e102-104.
[45] XU L, ZHAO G, YANG L, ZHU B, CHEN Y, ZHANG L, GAO X, GAO H, LIU G E, LI J. Genomic Patterns of Homozygosity in Chinese Local Cattle . Scientific reports, 2019, 9(1): 16977.
[46] FERENCAKOVIC M, HAMZIC E, GREDLER B, SOLBERG T R, KLEMETSDAL G, CURIK I, SOLKNER J. Estimates of autozygosity derived from runs of homozygosity: empirical evidence from selected cattle populations . Journal of Animal Breeding and Genetics, 2013, 130(4): 286-293.
[47] MASTRANGELO S, SARDINA M T, TOLONE M, DI GERLANDO R, SUTERA A M, FONTANESI L, PORTOLANO B. Genome-wide identification of runs of homozygosity islands and associated genes in local dairy cattle breeds . Animal, 2018, 12(12): 2480-2488.
[48] GOMEZ-RAYA L, RODRIGUEZ C, BARRAGAN C, SILIO L. Genomic inbreeding coefficients based on the distribution of the length of runs of homozygosity in a closed line of Iberian pigs. Genetics Selection Evolution, 2015, 16(47): 81.
[49] SHI L Y, WANG L G, LIU J X, DENG T Y, YAN H, ZHANG L C, LIU X, GAO H M, HOU X H, WANG L X, ZHAO F P. Estimation of inbreeding and identification of regions under heavy selection based on runs of homozygosity in a Large White pig population . Journal of Animal Science and Biotechnology, 2020, 28(11): 46.
[50] BOSSE M, MEGENS H J, MADSEN O, PAUDEL Y, FRANTZ L A, SCHOOK L B, CROOIJMANS R P, GROENEN M A. Regions of homozygosity in the porcine genome: consequence of demography and the recombination landscape . PLoS Genetics, 2012, 8(11): e1003100.
[51] GROENEN M A, ARCHIBALD A L, UENISHI H, TUGGLE C K, TAKEUCHI Y, ROTHSCHILD M F, ROGEL-GAILLARD C, PARK C, MILAN D, MEGENS H J,. Analyses of pig genomes provide insight into porcine demography and evolution. Nature, 2012, 491(7424): 393-398.
[52] HERRERO-MEDRANO J M, MEGENS H J, GROENEN M A, RAMIS G, BOSSE M, PEREZ-ENCISO M, CROOIJMANS R P. Conservation genomic analysis of domestic and wild pig populations from the Iberian Peninsula . BMC Genetics, 2013, 30(14): 106.
[53] FERREIRA E, SOUTO L, SOARES A M V M, FONSECA C. Genetic structure of the wild boar population in Portugal: Evidence of a recent bottleneck . Mammalian Biology, 2009, 74(4): 274-285.
[54] ZHANG Z, ZHANG Q, XIAO Q, SUN H, GAO H, YANG Y, CHEN J, LI Z, XUE M, MA P, YANG H, XU N, WANG Q, PAN Y. Distribution of runs of homozygosity in Chinese and Western pig breeds evaluated by reduced-representation sequencing data . Animal Genetics, 2018, 49(6): 579-591.
[55] XIE R, SHI L Y, LIU J X, DENG T Y, WANG L G, LIU Y, ZHAO F P. Genome-Wide Scan for Runs of Homozygosity Identifies Candidate Genes in Three Pig Breeds . Animals (Basel), 2019, 9(8): 518.
[56] PURFIELD D C, MCPARLAND S, WALL E, BERRY D P. The distribution of runs of homozygosity and selection signatures in six commercial meat sheep breeds. PLoS One, 2017, 12(5): e0176780.
[57] 刘家鑫, 魏霞, 邓天宇, 谢锐, 韩建林, 杜立新, 赵福平, 王立贤. 绵羊全基因组ROH检测及候选基因鉴定 . 畜牧兽医学报, 2019, 50(8): 1554-1566.
LIU J X, WEI X, DENG T Y, RUI R, HAN J L, DU L X, ZHAO F P, WANG L X. Genome-wide scan for run of homozygosity and identification of corresponding candidate genes in sheep populations. Acta Veterinaria et Zootechnica Sinica, 2019, 50(8): 1554-1566. (in Chinese)
[58] MASTRANGELO S, CIANI E, SARDINA M T, SOTTILE G, PILLA F, PORTOLANO B, BI.OV. ITA C. Runs of homozygosity reveal genome-wide autozygosity in Italian sheep breeds . Animal Genetics, 2018, 49(1): 71-81.
[59] MASTRANGELO S, TOLONE M, SARDINA M T, SOTTILE G, SUTERA A M, DI GERLANDO R, PORTOLANO B. Genome-wide scan for runs of homozygosity identifies potential candidate genes associated with local adaptation in Valle del Belice sheep . Genetics Selection Evolution, 2017, 49(1): 84.
[60] BEYNON S E, SLAVOV G T, FARRE M, SUNDUIMIJID B, WADDAMS K, DAVIES B, HARESIGN W, KIJAS J, MACLEOD I M, NEWBOLD C J, DAVIES L, LARKIN D M. Population structure and history of the Welsh sheep breeds determined by whole genome genotyping . BMC Genetics, 2015, 20(16): 65.
[61] FLEMING D S, KOLTES J E, MARKEY A D, SCHMIDT C J, ASHWELL C M, ROTHSCHILD M F, PERSIA M E, REECY J M, LAMONT S J. Genomic analysis of Ugandan and Rwandan chicken ecotypes using a 600 k genotyping array . BMC Genomics, 2016, 26(17): 407.
[62] ZHANG M, HAN W, TANG H, LI G, ZHANG M, XU R, LIU Y, YANG T, LI W, ZOU J, WU K. Genomic diversity dynamics in conserved chicken populations are revealed by genome-wide SNPs . BMC Genomics, 2018, 19(1): 598.
[63] PRYCE J E, HAILE-MARIAM M, GODDARD M E, HAYES B J. Identification of genomic regions associated with inbreeding depression in Holstein and Jersey dairy cattle . Genetics Selection Evolution, 2014, 46(1): 71-84.
[64] SAURA M, FERNANDEZ A, VARONA L, FERNANDEZ A I, DE CARA M A, BARRAGAN C, VILLANUEVA B. Detecting inbreeding depression for reproductive traits in Iberian pigs using genome-wide data . Genetics Selection Evolution, 2015, 47(1): 1.
[65] 史良玉, 王立刚, 张鹏飞, 莫家远, 李洋, 王立贤, 赵福平. 不同来源大白猪总产仔数近交衰退评估 . 畜牧兽医学报, 2021, 52(10): 2772-2782.
SHI L Y, WANG L G, ZHANG P F, MO J Y, LI Y, WANG L X, ZHAO F P. Evaluation of Inbreeding Depression on the Total Numbers of Piglets Born in Different Groups of Large White Pigs. Acta Veterinaria et Zootechnica Sinica, 2021, 52(10): 2772-2782. (in Chinese)
[66] NANI J P, PENAGARICANO F. Whole-genome homozygosity mapping reveals candidate regions affecting bull fertility in US Holstein cattle . BMC Genomics, 2020, 21(1): 338.
Advance in Genome-Wide Scan of Runs of Homozygosity in Domestic Animals
ZHANG PengFei1, SHI LiangYu1, LIU JiaXin1, LI Yang1, WU ChengBin2, WANG LiXian1, ZHAO FuPing1
1Key laborary of Animal Genetics Breeding and Reproduction (poultry), Ministry of Agriculture, Institute of Animal Science, Chinese Academy of Agricultural Sciences, Beijing 100193;2Quarantine Station of Animal Health Supervision and Administration Bureau in Maochikou Town, Changping District, Beijing 102202
Runs of homozygosity (ROH) is a long tract of homozygous genotypes commonly found in individuals and populations, which generates on the offspring’s genome inherited identical haplotypes from each parent. ROH contains a wealth of genetic information about populations, which makes it a useful tool for providing information to study how populations change over time. Moreover, ROH can estimate the genetic relationships between individuals to minimize the inbreeding mating rates. In addition, ROH can expose harmful mutations in the genome. The frequencies, sizes and distributions of ROHs in the genome are influenced by natural and artificial selection, recombination, linkage disequilibrium, population history, mutation rate and inbreeding level. Recently, with the use of high-throughput genotype technology and the reduction of second-generation sequencing costs, livestock and poultry breeding have entered into the genomic era. The selection intensity of the elites in livestock and poultry significantly increase to improve their performances, but it will increase inbreeding and cause inbreeding depression as well. Based on ROH molecular information, it is more accurately to detect past and nearest in close relative mating. The ROH-based inbreeding coefficient (F) can obtain an individual's true inbreeding coefficient,the realized inbreeding coefficient, and the pedigree-basedFis the expectation value of inbreeding coefficient. In the absence of genealogical information,Fcan be used to infer information about a group's history and the inbreeding levels. Meanwhile, the selection reshapes ROH patterns in different regions of the genome. In addition, the selection can increase the homozygosities around the target point, and harmful mutations are thought to occur more frequently in the ROH region, which can be detected by ROH to reduce the risk of complex diseases. After long-term selection, one ROH appeared in multiple individuals’ genomes in the same population, resulting in ROH islands. It has confirmed the correlation between ROH and the selected genomic region. The candidate genes related to economic traits can be annotated on the ROH islands by means of biological information. In addition, ROH also provides a new perspective for assessing the genetic diversity in domestic animals. Genome-wide ROH detection on the population can used to investigate the genetic structure of this population, andFcan evaluate the impact of inbreeding in the current breeding program, which can adjust breeding plans to protect the genetic diversity of varieties. Therefore, ROH has gradually become an important index to explore the historical population structure, the level of inbreeding, candidate gene identification. There are mainly two kinds of methods to identify ROH: observation genotype counting method and model-based analysis. Commonly used softwares include PLINK, GERMLINE, BEAGLE, GARLIC, etc. In practical applications, PLINK is the most common ROH detection tool. Since the SNP chip for cattle was firstly used in domestic animals, the cattle population was firstly conduct genome-wide ROH detection. Now, studies on ROH are becoming more popular in pigs, sheep and other domestic animals. This review mainly described the principle of ROH formation and its detection methods, as well as progress of its application in livestock and poultry, so as to provide reference for the genetic breeding of livestock and poultry.
runs of homozygosity; livestock and poultry breeding; single nucleotide polymorphism; inbreeding evaluation; candidate genes identification

2020-11-13;
2021-01-06
国家自然科学基金(31572357)、国家生猪产业技术体系(CARS-35)、中国农业科学院创新工程(ASTIP-IAS02)
张鹏飞,Tel:17806243706;E-mail:zhangpengfei3236@163.com。通信作者王立贤,E-mail:iaswlx@263.net。通信作者赵福平,E-mail:zhaofuping@caas.cn
(责任编辑 林鉴非)
