超低温SiO2 材料脆性开裂过程分子动力学模拟技术研究
2022-01-15沈才华李东彪李雪松郭金勇
朱 磊 沈才华 李东彪 李雪松 陈 伟 郭金勇
(1.中交南京交通工程管理有限公司, 南京 211800;2.河海大学 土木与交通学院, 南京 210098)
分子模拟方法是以计算机程序和模拟算法为工具在分子层面来研究物质的各种理化性质,再利用统计学总结出模拟体系的宏观物理量.分子动力学方法(molecular dynamic,MD)是其中一种非常重要、应用广泛的分子模拟方法[1].早在1957 年,Alder和Wainwright采用硬球模型,利用分子动力学研究气体和液体的状态方程,之后,学者们开始了大量的分子动力学研究[2].近年来分子动力学广泛应用于材料科学、无线电电子学、有机化学等领域[3-5].分子动力学模拟方法可以通过构造比较理想的分子构型模型,定量地再现真实固体中所发生的动态过程,但鲜有人应用分子动力学模拟SiO2材料脆性开裂过程.刘青松[6]基于Reax FF 反应力场,在微纳尺度下,应用大规模分子动力学方法,模拟了非晶态SiO2块体结构和非晶态SiO2薄片孔洞结构的单轴拉伸变形-断裂过程;研究了微观结构及尺寸对非晶态SiO2力学特性的影响,揭示了结构的微观变形机理.韩强等[7]运用分子动力学方法模拟了含有中心裂纹的扶手椅型石墨烯的拉伸破坏行为.谭旺[8]运用分子动力学方法对SiC/Al复合材料的界面力学性能和界面结构进行模拟研究分析,模拟结果显示塑性变形过程和断裂机制为:首先位错从界面发射,并向Al内部运动,遇到界面发生反射,随后发生滑移,并且有发生旋转和晶体重构的现象,滑移后有空洞生成,最后空洞增大形成裂纹而最终断裂.杨利等[9]用分子动力学方法,研究了预制边界裂纹并加入Nb的单晶γ-TiAL合金的裂纹扩展过程,分析了裂纹在1K 温度下的扩展行为.马磊等[10]采用分子动力学方法,结合Tersoff势函数,模拟了α-SiO2晶体的力学性能,并研究了温度对α-SiO2力学性能的影响.可见,随着分子模拟方法理论的完善,越来越多的学者开始采用分子动力学对不同材料的力学行为进行模拟探究,特别为揭示宏观现象下的微观机理提供了一种很好的途径.考虑不同工程荷载特点下裂纹扩展过程的研究较少,因此本文采用分子动力学模拟技术探究岩石材料中常见的SiO2材料脆性开裂的过程,建立断裂因子、微观参数、能量等裂纹扩展过程中微宏观的关系,为揭示固体材料裂纹扩展的微观机理研究提供参考,这对促进建筑材料的脆性开裂过程微观机理研究具有重要意义.
1 基于分子动力学的微观裂纹扩展模拟方法研究
分子动力学方法可以利用计算机模拟进行较大体系的长时间尺度的动态问题的研究,该方法特别适用于微纳米裂纹及扩展的研究.利用该方法不但可以观察到裂纹扩展中原子键断裂的细节、裂纹扩展的形式,还可计算出原子的位置和速度进而确定模拟中原子的运动轨迹、裂纹的扩展速度、体系的总势能和动能等物理量[11].
LAMMPS(Large-scale Atomic/Molecular Massively Parallel Simulator)是一款开源软件,由美国能源部的桑迪亚国家实验室(Sandia National Laboratories)研发,该软件免费且功能强大,不足之处是没有像其他商业软件的前处理与后处理用户界面,使用起来可能带来不便,因此LAMMPS的建模方法有:使用LAMMPS 的代码建模;借助其他软件进行建模.
LAMMPS对所含成分比较少的物质的纳观行为有较好的模拟效果,但是岩石是复杂混合体多孔结构,目前没有与之一一对应的势函数可以使用,所以本文采用岩石材料中最常见且分子构型较简单的二氧化硅(SiO2)来分析岩石基材的裂纹扩展过程,探索建立工程材料的纳观机理分析方法.
分子动力学模拟方法的原理是通过原子间的相互作用势,按照经典牛顿运动定律求出原子轨迹及其演化过程.因此,建立合适的势能函数是进行分子动力学模拟的第一步,也是最关键的一步.势能函数的正确与否,直接关系到模拟结果的精确性和可靠性[12].
本文SiO2的模拟采用的是vashishta势函数[13],表达式为:


表1 Si-O-O 参数表[12]
二氧化硅(SiO2)的模拟采用的建模方法是第二种方法,使用VMD 软件建立SiO2模型,该软件为一种分子可视化程序,是Visual Molecular Dynamics的缩写,由University of Illinois at Champaign的Theoretical Biophysics Group研发(具体分子构型如图1所示),建模过程如图2所示,尺寸为:x·y·z=199.16×99.56×6.948Å.
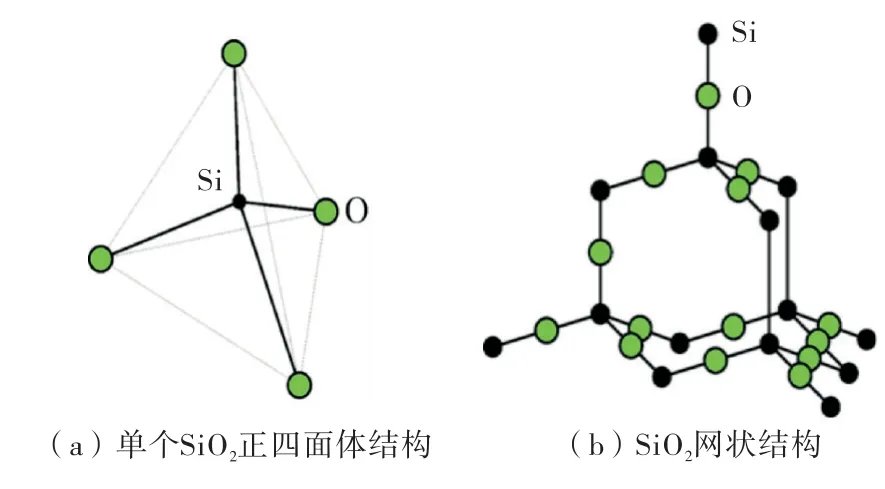
图1 二氧化硅空间构型

图2 计算模型示意图
2 NVE系综裂纹扩展行为分析
初始模型温度为10 K,初始裂纹长度10Å,采用消除A 和B交界面的作用模拟初始裂缝,即不考虑初始裂缝宽度和形状.位移加载速度为10Å/ps,模拟分析裂纹扩展过程.模拟结果显示:在拉伸过程中,能量逐渐向裂纹尖端聚集,裂纹尖端出现应力集中,分子之间吸引力较小的部位慢慢被拉开,直至势函数作用距离之外,尖端开裂.如图3(a)所示,在加载10 ps左右,裂纹尖端出现空位,显然,该空位没有出现在裂纹正前方,是二氧化硅的构型决定的,即原子的空间排布导致空位处的原子之间势能较低.加载到20 ps,如图3(b)所示,预制裂纹尖端及空位各自形成一个裂纹尖端,即“裂纹1”和“裂纹2”(空位处),随后,在两个裂纹尖端的“竞争”中,裂尖之间的原子被拉开直至拉断,“裂纹1”和“裂纹2”合并后继续向前扩展,如图3(c)、(d)所示.图3(d)、(e)、(f)分别表示裂纹扩展至图2(c)所示的“圆1”、“圆2”和“圆3”的位置.
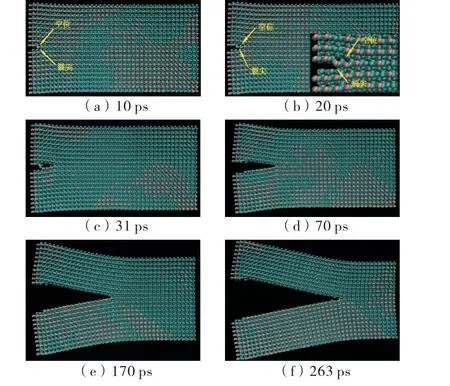
图3 裂纹扩展过程
在裂纹扩展路径,即拉伸方向上取11个贴近断裂面的点(图4(a)),间距为20Å×10=200Å.观察这些点y方向位移变化(图4(b)),判断各点起裂时间,并计算裂纹扩展的大致速度以及裂纹长度变化(图4(c)、(d)).由图4(c)可看出,裂纹在扩展过程中并不是匀速前进的,而是上下震荡.
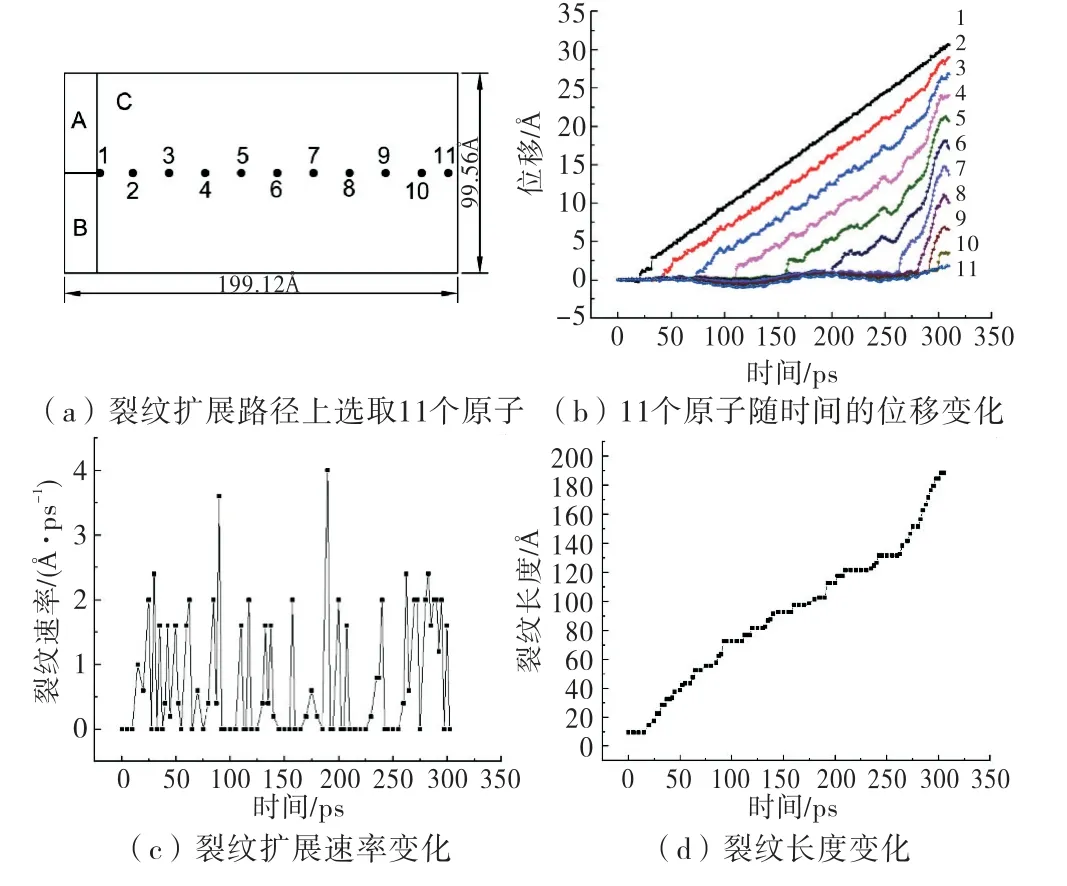
图4 裂纹扩展速率与长度
结合图4(d)可看出,裂纹长度曲线斜率在260 ps之前逐渐变小,之后曲率增大.所以速率变化的总体趋势是先减小(260 ps之前)后增大.不计裂纹停止的时间,裂纹扩展速率在2Å/ps(200 m/s)左右,最高速率超过4Å/ps(400 m/s),该计算结果与S.Chandra等人[14]计算的Al在1K 温度下扩展速率320 m/s比较接近,所以本模型计算结果基本合理可靠.
在加载过程中,图2(c)的A 区域的平均应力如图5(a)所示,整个过程应力变化为先减小后略微增大,从260 ps左右开始再次减小.应力的计算采用的是维里应力[14],维里应力表征原子间相互作用力,可正可负,表达式为:
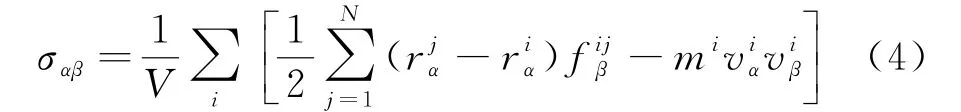
式中:V是需要计算应力部分的体积;i,j表示原子i周围的j原子;r iα,r jα表示i原子和j原子在α方向的位置;f ijβ表示β方向j原子对i原子的力;m i表示i原子的质量;v iα和v iβ分别表示i原子在α和β方向的速度.
初始应力较高的原因是,在裂纹扩展前,A 区域的右下角处于预制裂纹尖端的附近,该处应力集中,从而导致A 区域总体平均应力较大.裂纹扩展后,裂纹尖端逐渐远离A 区域,因此该区域应力被释放后下降,且应力分布逐渐均匀.应力峰值时刻对应裂纹加速扩展时刻.图5(b)裂纹应力强度因子按照断裂力学应力强度因子公式来计算:

式中;KⅠ是裂纹应力强度因子;Y是与裂纹形状和加载方式有关的系数[15],这里取1.12;σ是A 区域平均应力,取图5(a)中的数据;c是裂纹总长度,取4(d)中的数据.应力强度因子计算结果如图5(b)所示,应力强度因子逐渐升高,20 ps左右裂纹起裂,此时裂纹应力强度因子可能达到了断裂韧性(根据图5(a)计算的A 区域平均最大应力为380 MPa,初始裂纹长度为10,代入公式(5)可获得其断裂韧度KⅠC=0.02 MPa·m1/2),导致裂纹开裂.裂纹驱动力的表达式为:
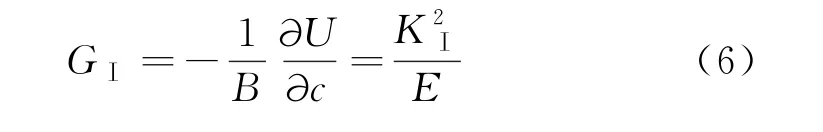
其中:GⅠ是裂纹驱动力;B是模型宽度;-∂U/∂c是指系综总势能随着裂纹扩展降低的量;E是弹性模量.20 ps左右裂纹起裂.据资料可得[16]:二氧化硅弹性模量约为70 GPa,计算得到起裂裂纹驱动力为0.005 7 J/m2.
如图6所示,裂纹失稳扩展对应图中的切点,c>c0时有∂C/∂c>∂R/∂c,即裂纹扩展动力增长率高于阻力增长率,裂纹失稳扩展.本文理论曲线取自《应力强度因子手册》[17].由图5(c)可看出,理论曲线与计算结果在虚线之前吻合较好,虚线之后开始偏离,可能是加载方式以及模型尺寸效应导致.当裂纹驱动力GⅠ达到最大值,对应图5(b)中应力强度因子的KⅠmax处,从该处开始,应力强度因子下降,亦即裂纹驱动力下降,有∂G/∂c<∂R/∂c,裂纹停止扩展,此时对应图6中G曲线的顶点.在裂纹驱动力(阻力)曲线图中,可大致分为Ⅰ、Ⅱ、Ⅲ3个阶段,Ⅰ阶段∂G/∂c<∂R/∂c,裂纹阻力大于驱动力,裂纹未扩展,Ⅱ阶段∂G/∂c>∂R/∂c,裂纹驱动力大于阻力,裂纹扩展,Ⅲ阶段∂G/∂c<∂R/∂c,裂纹扩展动力不足,停止扩展.
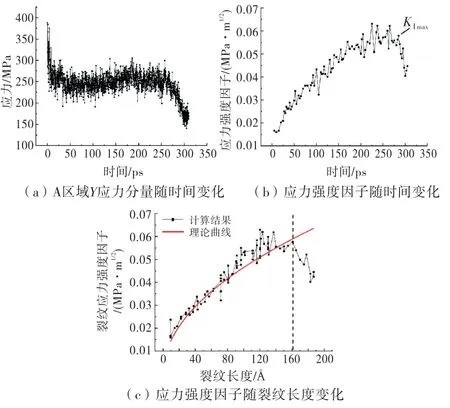
图5 A 区域应力与裂纹应力强度因子
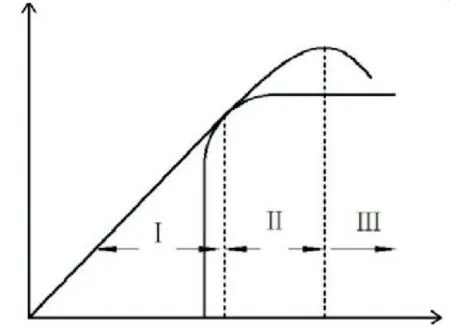
图6 裂纹扩展动力-阻力曲线
在裂纹扩展过程中,图2(c)中3个圆形裂纹尖端区域能量变化如图7(a)所示,3个区域变化趋势大致相同,在裂纹扩展的瞬间能量突然升高,随后维持稳定,而且NVE 系综无其他能量耗散项,因此可认为能量升高是因为3个区域裂纹断裂后的表面储存有表面能.从图7(b)中可看出,C 区域系统动能变化不明显,可以认为3个圆形裂纹尖端区域能量升高的部分均是表面能,每个圆形区域裂纹开裂截面面积为41.688Å2,开裂后能量增长约为0.6 e V,所以表面能为0.23 J/m2.计算过程是在极低温度(10 K)下进行,而且断裂过程裂纹尖端始终是尖锐的,所以模型高度脆性.Lawn[18]给出的高度脆性材料的表面能在10-1~10之间,本模型计算的表面能结果在该范围内,说明本方法模拟分析结果基本合理.
由图7(a)可计算出裂纹从圆1扩展到圆2的速率v12=0.49Å/ps,以及从圆2扩展到圆3的平均速率v23=0.46Å/ps.
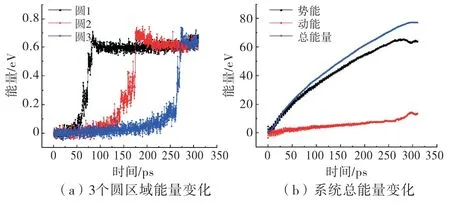
图7 能量变化
由裂纹尖端应力场的弹性解表达式可知,距离裂纹尖端越近,应力越大.理论上,裂纹尖端应力趋于无穷大,这是不符合物理规律的,所以裂纹尖端应力到底多大时裂纹开裂是弹性力学解无法触及的“黑匣子”.物理层面,裂纹尖端是由原子分布形成的,图3已经说明了裂纹扩展是原子之间相互作用断开即键断裂导致的,所以裂纹尖端应力达到一定程度,原子键被拉断,裂纹扩展.为了探究裂纹尖端应力变化,得到裂纹扩展过程,计算图2(c)中圆1、2、3三个区域水平和竖直方向应力变化,如图8所示.在微观尺度上,原子时刻都在运动,因此应力值是波动的.图8(a)是3个裂纹尖端区域水平方向应力的变化,裂纹依次进入3个区域,3个区域先后出现峰值,峰值对应裂纹进入该区域的时间,可见裂纹扩展过程尖端水平向应力最大可达到4 GPa,同理,8(b)表示裂纹扩展过程中3个尖端区域竖直向应力变化,裂纹进入的瞬间应力可高达6 GPa左右.对比8(a)和8(b)可发现,水平向应力是逐渐增加、逐渐降低,垂直向则是快速增加、裂纹扩展后几乎是瞬间下降至裂纹未靠近的应力值范围,说明裂纹尖端在向前推移的过程中,水平向应力受影响的原子远多于垂直向.裂纹扩展时,裂纹尖端应力的“奇异性”是不存在的,应力增加到6 GPa左右裂纹会裂开并向前扩展.
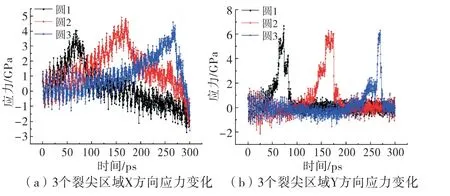
图8 裂纹扩展过程裂尖应力变化
3 不同系综对裂纹扩展行为的影响规律
分子动力学模拟,总是在一定的系综下进行的,不同系综反映了不同的工程环境,实际工程环境非常复杂,进行机理研究时一般假设为不同的理想环境,即用不同的系综来描述.系统原子数N,体积V,能量E保持不变的环境称为NVE 系综.系统原子数N,体积V,温度T保持不变,且总动量保持不变,则称为NVT 系综.在NVT 系综下进行计算,计算结果如图9所示.
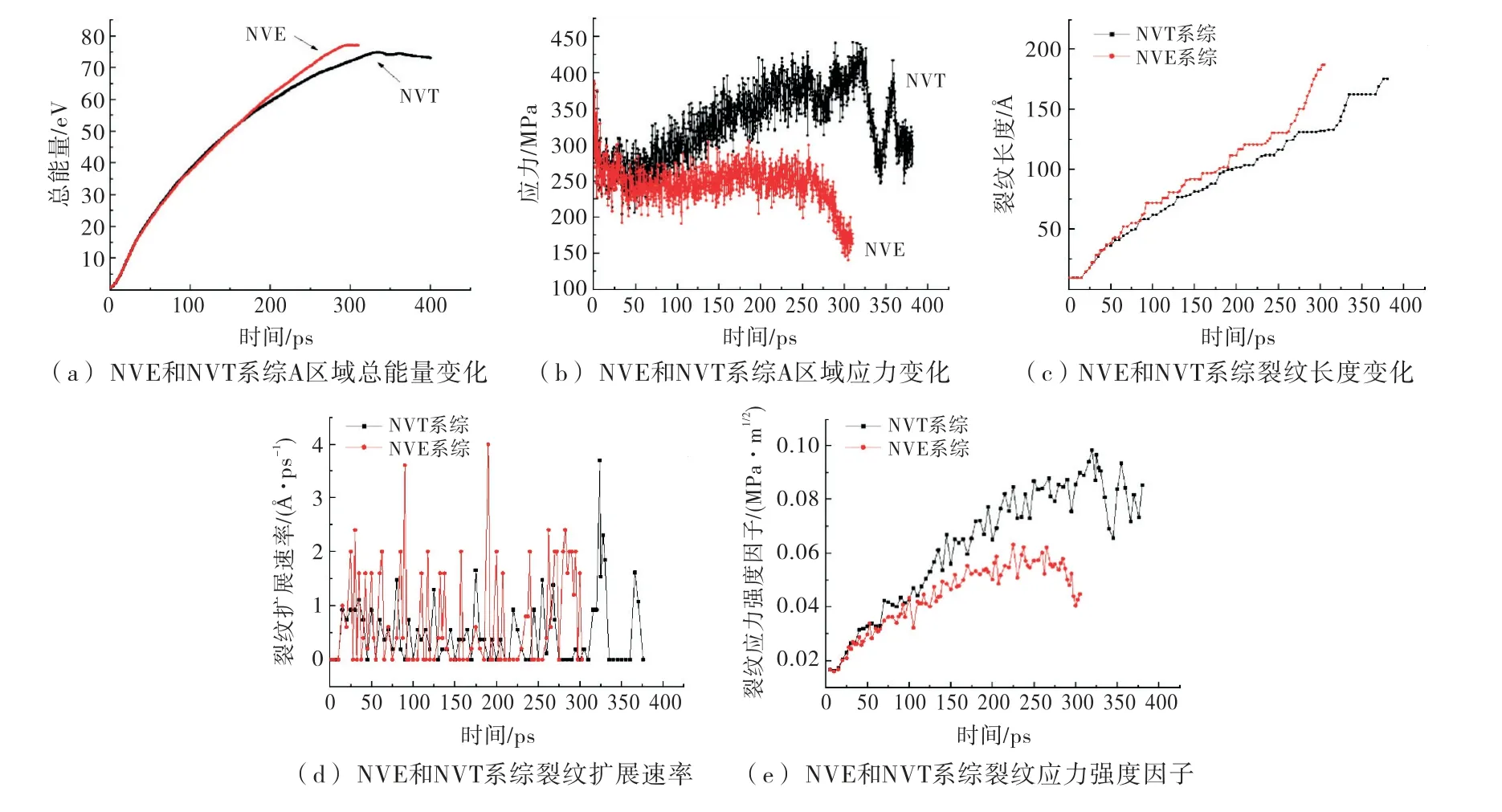
图9 NVT 系综下的计算结果
从如9(a)可看出,C区域NVT 总能量增长幅度小于NVE 系综,分析认为NVT 系综可能与外界环境有能量交换.从图9(b)可看出,两个系综A 区域Y方向应力开始时刻相似,随后,NVT 系综下应力增大,NVE系综基本不变,最后二者又都经历减小的过程.由图9(c)、(d)可看出,NVE系综下裂纹扩展平均速率高于NVT 系综,从图9(e)可看出,NVT 系综下,裂纹扩展的应力强度因子大于NVE 系综下的计算结果,这可能是由于NVT 系综允许与外界能量交换,能量损失较大,导致裂纹扩展需要更大的裂纹应力强度因子.
4 结 论
宏观计算在一定程度上规避了裂纹尖端的力学行为,而裂纹尖端的力学行为恰恰是裂纹扩展研究的关键.裂纹的起裂扩展归根结底是材料微观尺度上原子之间相互作用力从有到无的过程,即键断裂的过程.采用vashishta势函数,使用VMD 软件的建立SiO2模型,使用开源分子动力学软件LAMMPS 对岩石常见主要成分SiO2进行微观尺度裂纹扩展行为研究,不仅计算结果和呈现规律基本合理,而且可以很好地揭示微观系统对裂纹扩展过程的影响,为探究工程材料的破坏微观机理提供了一种新途径.本文在建立了分子动力学模拟技术模拟SiO2材料裂纹过程微观机理研究方法的基础上,针对NVE 和NVT 系综下对裂纹扩展的行为规律也做了对比,其主要结论如下:
1)在拉伸过程中,能量逐渐向裂纹尖端聚集,裂纹尖端出现应力集中,势能较小的原子之间慢慢被拉开,裂纹扩展方向受分子构型影响,并非直线,尖端拉裂的过程非常短暂,计算的拉裂强度最高达6 GPa,而且裂纹扩展过程中计算应力值呈现波动状态.裂纹扩展可大致分为Ⅰ、Ⅱ、Ⅲ3个阶段:Ⅰ阶段∂G/∂c<∂R/∂c,裂纹阻力大于驱动力,裂纹未扩展;Ⅱ阶段∂G/∂c>∂R/∂c,裂纹驱动力大于阻力,裂纹扩展;Ⅲ阶段∂G/∂c<∂R/∂c,裂纹扩展动力不足,停止扩展.
2)不同系综下裂纹扩展速率是不同的,NVE 系综下裂纹扩展平均速率高于NVT 系综;NVT 系综允许与外界能量交换,裂纹扩展的计算应力强度因子大于NVE系综的计算值.
本文方法能有效反映不同环境下裂纹微观扩展机理的区别,并揭示其断裂因子和能量的变化规律,分析结论基本符合实际情况,为探究其他常见岩石类材料或工程材料的微观机理研究提供了新的途径.
