青稞秸秆灰掺入氯氧镁水泥中的活性与作用机理*
2022-01-14乔宏霞舒修远
曹 锋,谭 镇,乔宏霞,舒修远
(1.青海民族大学 土木与交通工程学院, 西宁 810000;2.兰州理工大学 土木工程学院, 兰州 730050)
0 引 言
青稞是一种适宜生长在青藏高原寒冷环境的谷类作物,青海省青稞种植面积约占全国青稞总种植面积的41.71%[1]。目前,青稞秸秆主要被当地农牧民用作燃料和牲畜饲料,但是青稞秸秆中灰分含量较高,尤其灰分中二氧化硅含量较高,并不非常适合作为家用燃料和牲畜饲料[2]。为了保护生态环境,禁止在农田焚烧秸秆。此外,由于青藏高原气候寒冷,降雨量少,青稞秸秆的还田处理成效甚微。大量的秸秆资源堆积,使用效率低,对当地居民的生产生活造成了一定的影响。因此,对HBSA的活性性能进行研究,并进一步将其应用于建筑材料中,具有重要的应用价值。
国内外学者研究发现,粉煤灰[3-4]、稻壳灰[5-6]、秸秆灰[7-9]等材料由于具有火山灰效应,对水泥基材料的性能有显著的改善作用。张强等[10]研究了在马弗炉中500 ℃煅烧5 h所得油菜秸秆灰,以10%比例替代水泥时,混凝土的抗拉强度降低了25%,抗压强度仅下降了8%。Gomes等[11]发现在氯氧镁纤维水泥中添加稻壳灰,可以有效地改善其微观结构,提高其力学性能和耐久性。Qudoos等[12-13]发现麦秸灰的火山灰和填充效应使水泥基复合材料的微观结构更加致密,抗压强度提高。Cordeiro等[14]将甘蔗渣灰作为矿物掺合料掺入水泥砂浆中,发现抗压强度与灰的粒径和细度有着直接关系,较细的灰能产生更高的抗压强度和火山灰活性。然而,上述灰分能够提高水泥基材料性能的共同原因在于,均含有一定量的活性二氧化硅,与水泥水化产物进一步发生反应,生成对其性能有利的新的产物。因此,青稞秸秆灰分中较高的二氧化硅含量,使其作为水泥基辅助胶凝材料成为了可能。
然而,关于HBSA的活性制备条件以及对水泥混凝土材料性能的影响却未曾报道。本文将不同煅烧温度、煅烧时间及研磨时间下制备的HBSA,分别掺入到氯氧镁水泥砂浆(MOCM)中,通过宏观力学试验及微观结构测试确定HBSA的活性制备条件。进一步分析各影响因素的主次关系,最终揭示活性效应产生的机理。
1 实 验
1.1 实验原材料
轻烧氧化镁粉和工业氯化镁均为青海省格尔木市察尔汗盐湖氯化镁厂生产。轻烧氧化镁粉中MgO含量为98%,活性MgO含量为62.4%。工业氯化镁中MgCl2·6H2O含量为96%。砂子采用粒径小于4.75 mm的青海贵德河沙,级配良好。拌和用水采用自来水,符合混凝土拌和用水的质量标准。减水剂采用聚羧酸系高效减水剂,减水效率为21%。耐水剂采用磷酸,H3PO4的含量不小于85%,色度黑曾单位不大于25。青稞秸秆取自青海省互助县南门峡地区,去除杂草等杂质后,在室外自然环境下焚烧成灰分。将初次焚烧的HBSA去除泥土、砂砾等杂质后,在实验室条件下采用一体式SX2-12-10A智能箱式马弗炉进行二次煅烧。煅烧后的灰分待其冷却后,在辊式球磨机中进行机械研磨。
1.2 试件制备
氯氧镁水泥砂浆的配合比,见表1。将不同煅烧温度、煅烧时间、研磨时间条件制备的HBSA,以氧化镁质量15%的比例掺入到MOCM混合料中,拌合均匀后浇筑成40 mm×40 mm×160 mm的棱柱体试块,每组3块。室内自然条件下养护24 h后拆模,然后在自然条件下继续养护至28 d,养护温度为(20±2)℃,养护湿度不低于50%。
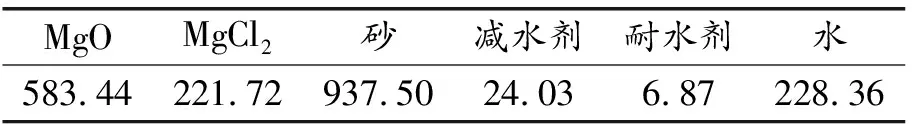
表1 氯氧镁水泥砂浆配合比/(kg/m3)
1.3 实验方法
对不同煅烧条件下的HBSA进行XRD测试,然后利用origin软件对XRD图谱进行分峰拟合并求解结晶峰面积及总峰面积,然后按照下式来计算XRD图谱的结晶度δ[15]:
(1)
式中:Ac为XRD图谱的结晶峰数学面积;At为XRD图谱的总峰数学面积。
采用水泥胶砂抗折抗压试验机对棱柱体试件进行抗折、抗压强度试验。根据标准GB/T 17671—1999的规定[16],棱柱体试件先进行抗折强度试验,折断后的半截棱柱体侧面上再进行抗压强度试验,取其每组3块试件测试的平均值作为强度测试结果。由于氯氧镁水泥的性能主要由氧化镁、氯化镁与水的摩尔比决定,且氯氧镁水泥无规范标准可参考,因此,相对活性指数测试参考标准JG/T 315-2011执行[17]。该实验中HBSA以外掺的方式掺入到MOCM中,并按照下式计算相对活性指数φ:
(2)
式中:fw为掺入HBSA的MOCM试件28 d抗压强度,MPa;fd为未掺HBSA的MOCM试件28 d抗压强度,MPa。
1.4 微观测试
采用布鲁克D8型X射线衍射仪(XRD)对不同煅烧温度及煅烧时间的HBSA进行物相分析;采用Malvern激光粒度仪(LPSA)对不同研磨时间HBSA的粒径进行测试;采用X射线荧光光谱仪(XRF)对HBSA的化学组成进行定量分析;采用Regulus8100型场发射扫描电子显微镜(SEM)对MOCM进行微观形貌测试。HBSA的微观测试样品,取其煅烧、研磨后的粉末适量,过80 μm的筛去除其杂质后,分别进行XRD以及XRF测试。粒径测试样品取其研磨后的粉末直接进行测试。MOCM的SEM测试样品取MOCM试块进行力学性能试验后的薄片装样品,厚度不大于1 cm,浸入酒精48 h以终止其进一步水化,取出干燥后进行SEM测试。
2 结果与讨论
2.1 活性分析
煅烧温度为500、600、700 ℃,煅烧时间为5 h时HBSA的XRD图谱如图1所示,结晶度计算结果见表2。可以看出,不同煅烧温度时HBSA的主要成分为SiO2,但组成比例差异显著。煅烧温度为700 ℃的SiO2晶体衍射峰明显强于600和500 ℃时,且600 ℃时该峰最弱。500 ℃时HBSA的物相组成中,有明显的CaAl2Si2O8·4H2O衍射峰,而600、700 ℃时该物相衍射峰逐渐变弱。此外,由XRD结晶度计算结果可知,随着煅烧温度的增加,HBSA的XRD结晶度先减小后增加。煅烧温度为600 ℃时结晶度最小,其次为700,500 ℃结晶度最大。因此,600 ℃煅烧所得HBSA活性最高。
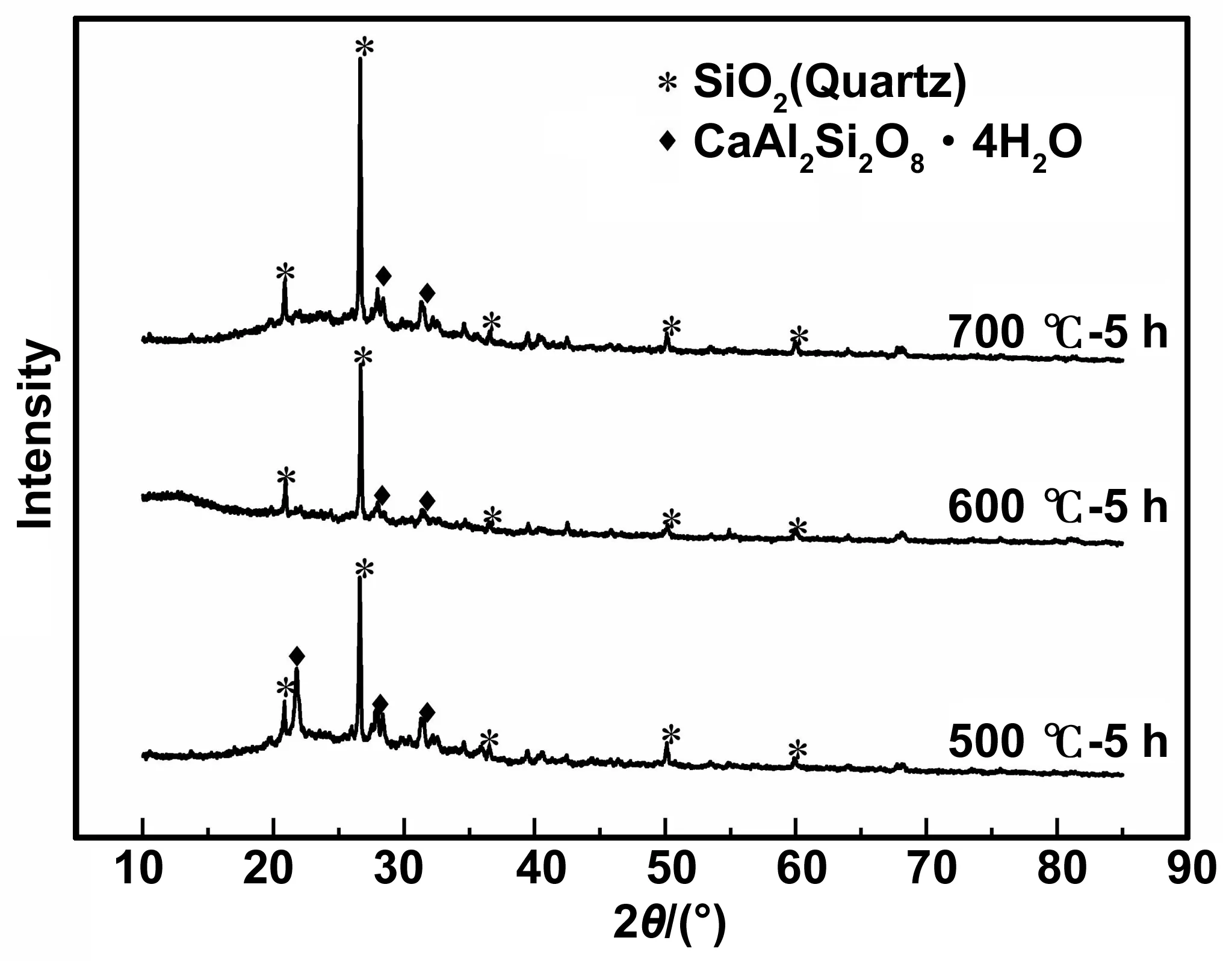
图1 不同煅烧温度HBSA的XRD图谱
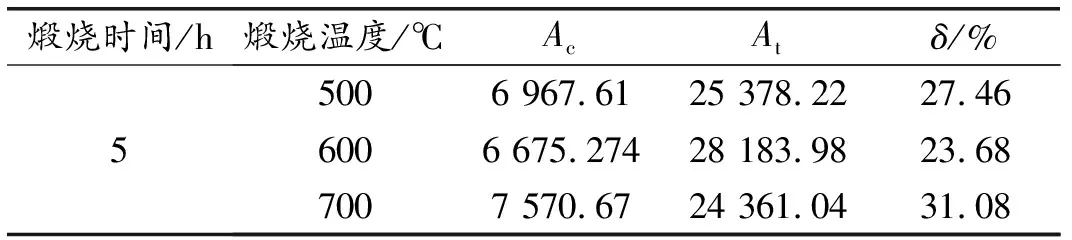
表1 不同煅烧温度HBSA的结晶度
煅烧温度为600 ℃、煅烧时间为2、5、8 h时HBSA的XRD图谱,如图2所示。XRD结晶度的计算结果见表3。可以看出,不同煅烧时间HBSA的物相种类基本一致,HBSA的主要成分为SiO2,但组成比例差异显著。煅烧时间为5和8 h的SiO2晶体衍射峰明显强于2 h,说明煅烧2 h时HBSA中所含晶体SiO2最少。因此,随着煅烧时间的增加,更多的无定型SiO2转变为晶体SiO2。由XRD结晶度计算结果可知,煅烧时间为2 h时结晶度最小,其次为煅烧5 h,煅烧8 h结晶度最大。因此,600 ℃煅烧温度下煅烧2 h所得HBSA活性最高。
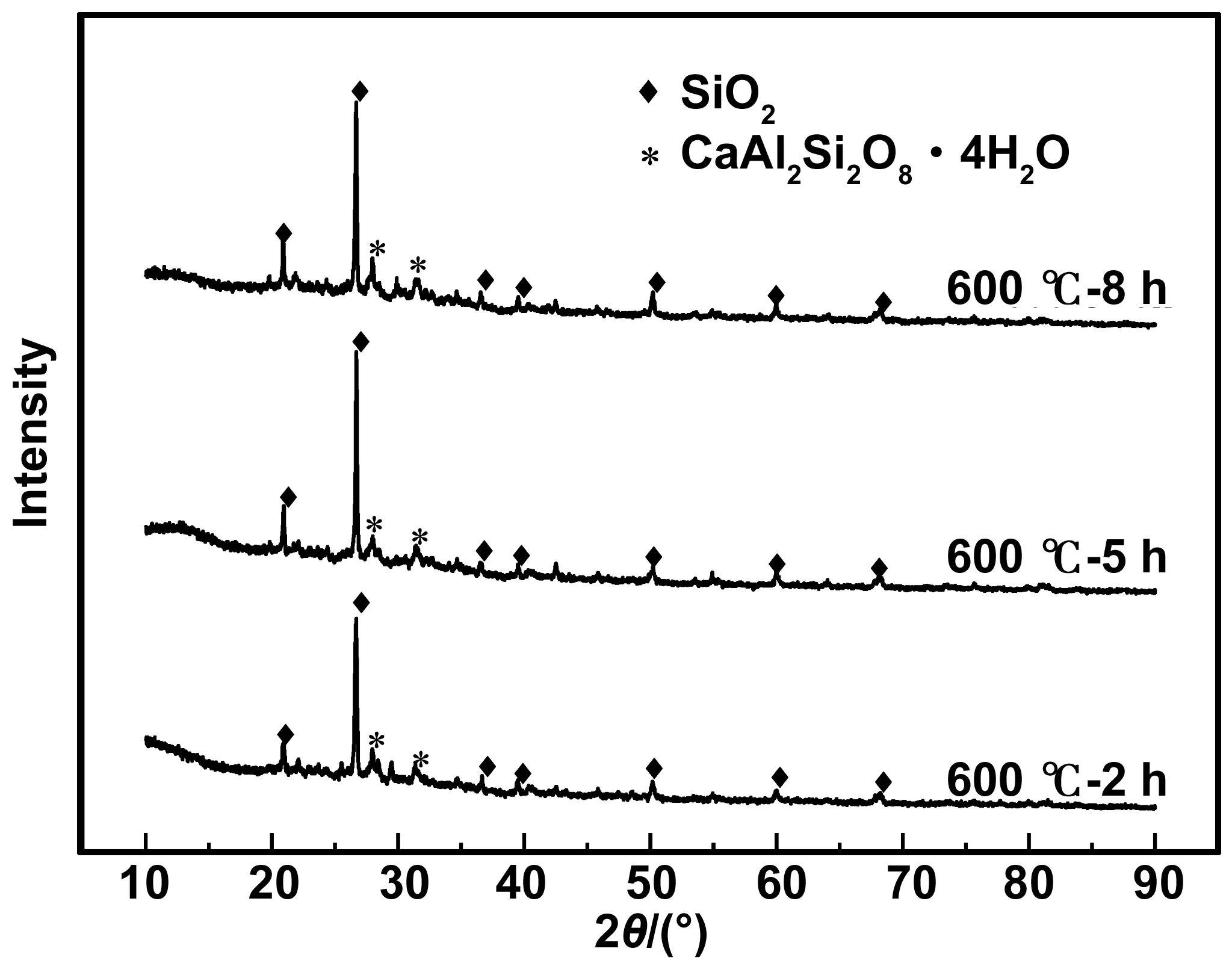
图2 不同煅烧时间的XRD图谱
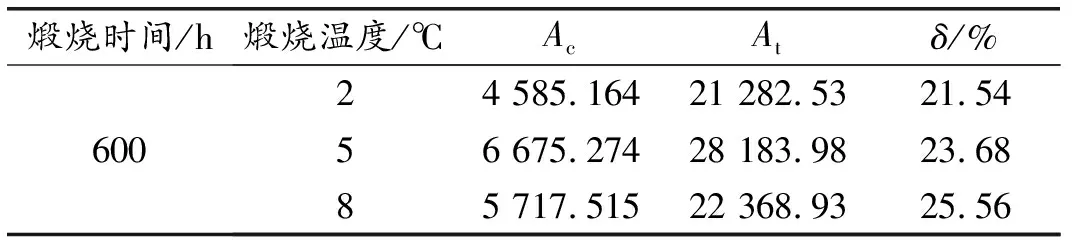
表3 不同煅烧时间HBSA的结晶度
HBSA研磨0、0.5、1、2、3 h的粒径测试结果,如图3所示。Sa45(45 μm方孔筛的筛余量)、平均粒径、比表面积等参数测试结果,见表4。可以看出,不同研磨时间的粒径分布显著不同。未研磨时,粒径分布曲线出现了一段水平线,说明该粒径分布具有明显的不均匀性,存在非连续的较大粒径分布情况。随着研磨时间的增加,粒径分布曲线逐渐向左移动,当研磨超过2 h以后,粒径分布曲线又开始向右移动。说明粒径先逐渐减小,随后又开始增大。相同累积通过率所对应的粒径,研磨2 h时最小,未研磨时最大。相同粒径所对应的累计通过率,研磨2 h时最大,未研磨时最小。由表4可以看出,随着研磨时间的增加,Sa45及平均粒径先减小后增加,比表面积先增大后减小。研磨2 h所得HBSA,Sa45的值最小,平均粒径最小,比表面积最大。与未研磨时相比,灰分的平均粒径减小为原来的4.21%,比表面积增加了2.77倍。研磨3 h时,平均粒径增大,比表面积减小。这是由于过度研磨导致灰分出现了轻微的团聚现象,从而使得试验所测粒径增大,比表面积减小。说明有效研磨时间应控制在2 h以内,研磨效果较好。
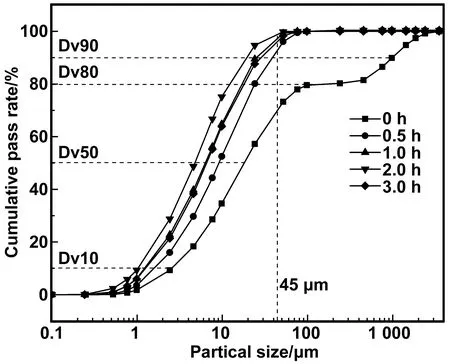
图3 不同研磨时间HBSA粒径分布
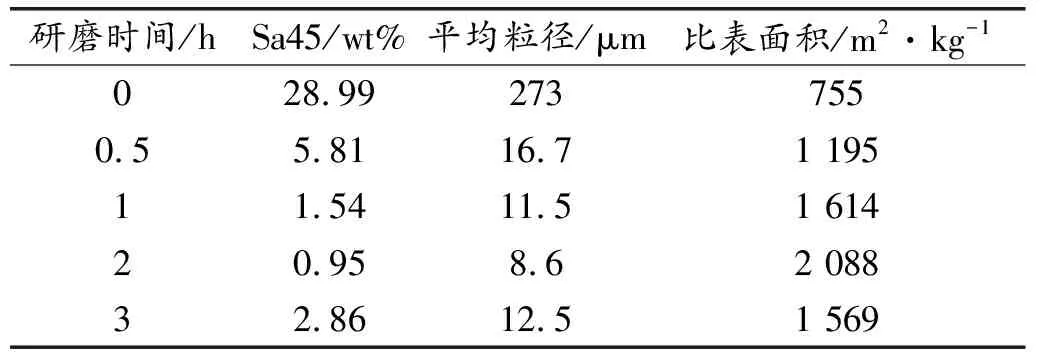
表4 HBSA的粒径分布参数
2.2 相对活性指数
煅烧温度为500、600、700 ℃,煅烧时间为2、5、8 h,研磨时间为0.5、1、2 h条件下所制备的HBSA,分别掺入MOCM中的抗压强度测试结果如图4所示。随着煅烧温度的增加,抗压强度先增大后减小,600 ℃时抗压强度最高,500 ℃次之,700 ℃最低。随着煅烧时间的增加,煅烧温度为500和600 ℃ 时抗压强度逐渐减小,而700 ℃抗压强度逐渐增大。说明500和600 ℃ 时,随着煅烧时间的增加,HBSA中的活性成分逐渐减少,进而导致其抗压强度逐渐减小。700 ℃ 时HBSA中活性成分较少,此时以晶体SiO2为主,主要发挥HBSA的颗粒填充效应。此外,500、600和700℃ 时,随着研磨时间的增加,抗压强度均随之增加。说明充分的研磨可以有效发挥HBSA的活性效应以及填充效应。
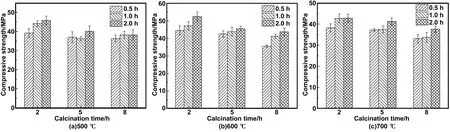
图4 掺入不同制备条件HBSA的MOCM的抗压强度
煅烧温度为500、600、700 ℃,煅烧时间为2、5、8 h,研磨时间为0.5、1、2 h条件下所制备的HBSA,分别掺入MOCM中的相对活性指数计算结果如图5所示。可以看出,随着煅烧时间的增加,500、600 ℃时相对活性指数逐渐减小,700 ℃ 时相对活性指数逐渐增加。而且,700 ℃ 时HBSA的活性低于500、600 ℃,600 ℃时活性最高。随着研磨时间的增加,500、600以及700 ℃下煅烧的HBSA活性指数均逐渐增加。说明在有效研磨时间内,研磨时间越长,相对活性指数越高。600 ℃煅烧2 h、研磨2 h所得HBSA掺入MOCM中的相对活性指数为1.06,达到了所有制备条件下HBSA相对活性指数的最大值。因此,600 ℃煅烧2 h、研磨2 h为HBSA的最佳制备条件。
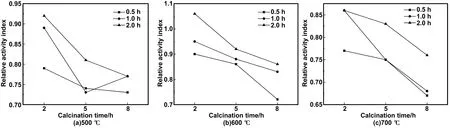
图5 掺入不同条件HBSA的MOCM的相对活性指数
根据上述分析,600 ℃时煅烧2 h、研磨2 h应为最佳制备条件,但是由于煅烧时间设置幅度较大,尚未分析2 h左右的煅烧时间是否更佳。因此,为了进一步确定HBSA的最佳制备条件,在上述分析的基础上,增加600 ℃时煅烧1 和3 h的测试结果,如图6所示。煅烧温度为600 ℃时,随着煅烧时间的增加,抗压强度及相对活性指数先增加后减小,煅烧2 h时达到最大,3 h次之,1 h最小。研磨2 h以内时,随着研磨时间的增加,抗压强度及活性指数逐渐增加。可见,有效的研磨可以增加其活性。此外,煅烧时间过长或过短,都会造成MOCM的相对活性指数减小,即HBSA的活性性能下降。因此,600 ℃煅烧2 h、研磨2 h所得HBSA的活性最好。
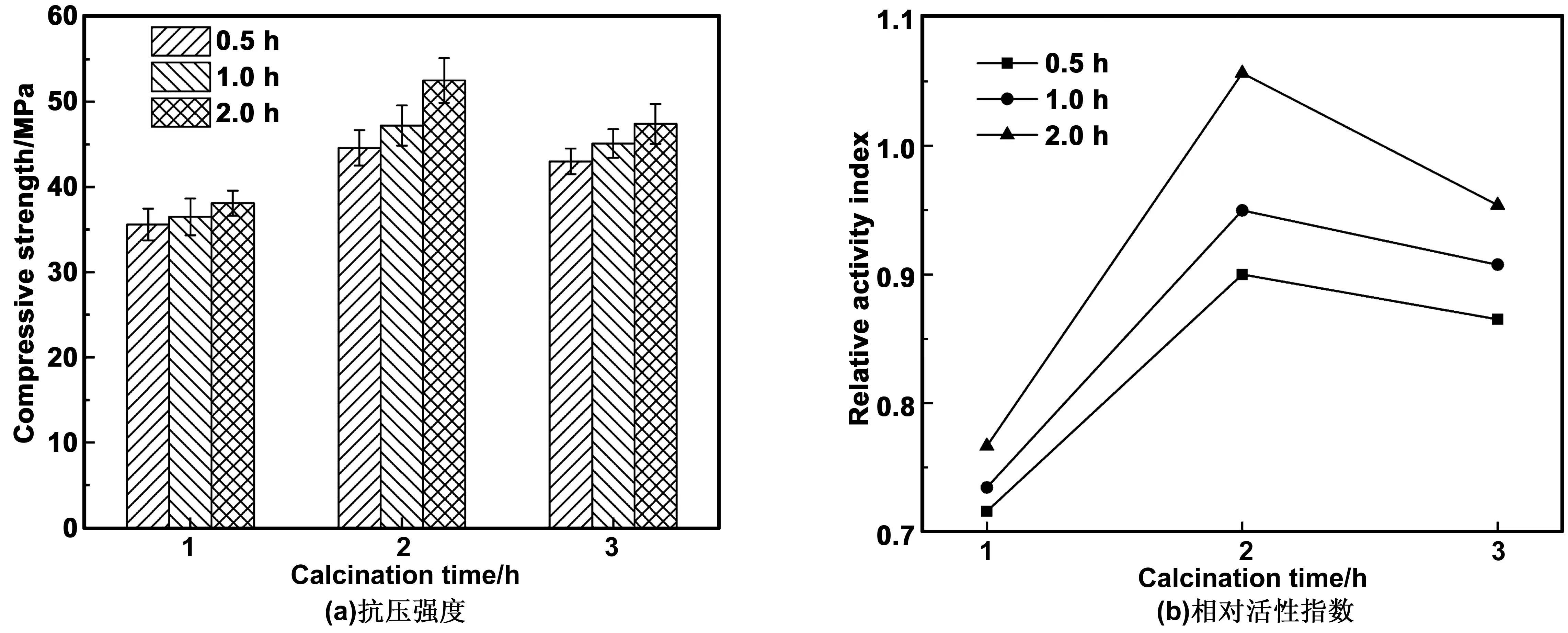
图6 MOCM的抗压强度及相对活性指数
2.3 灰熵关联分析
灰熵关联度分析是在灰色关联度分析的基础上引入信息熵理论,避免灰色关联分析灰关联度时局部关联度造成整个灰关联度倾向出现的损失,并对系统中的各个影响因素间的相似程度、吻合程度做定量描述[14]。对不同制备条件下的HBSA掺入MOCM中的抗压强度进行分析,以确定影响其活性的3个因素:煅烧温度、煅烧时间、研磨时间的主次关系,进而为HBSA的制备条件提供可靠的设计依据。
反映系统行为特征的数据序列为参考数列,影响系统行为的数据序列为比较数列[18]。掺入不同制备条件HBSA的MOCM抗压强度为参考数列,记为xθ(k),k=1,2,…,n;煅烧温度、煅烧时间、研磨时间等特征参数为比较数列,记为xi(k),k=1,2,…,n。上述不同因素下制备的HBSA对MOCM抗压强度的影响进行灰熵关联分析,并按下式计算灰熵关联系数δi(k),灰关联系数分布密度值ρh,灰关联熵H(ρh)以及灰熵关联度E(xi)[19]:
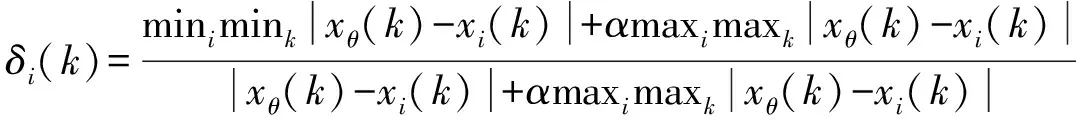
(3)
(4)
(5)
(6)
式中,α为灰色关联系数δi(k)的区分度系数,取0.5;Hmax为差异信息列的最大熵,取Hmax=ln(n)。
对上述各制备条件下HBSA的煅烧温度、煅烧时间、研磨时间作为比较数列,掺入各条件HBSA的MOCM的抗压强度作为参考数列,全部进行无量纲均值化处理。然后按照上式(3)~(6)计算灰熵关联分析相关参数,见表5。可以看出,煅烧温度、煅烧时间、研磨时间的灰熵关联度大小为:煅烧温度>研磨时间>煅烧时间。说明对MOCM中掺入HBSA的抗压强度影响最主要的因素为煅烧温度,其次为研磨时间,煅烧时间影响最弱。因此,制备活性HBSA时,应重点控制秸秆灰的煅烧温度。
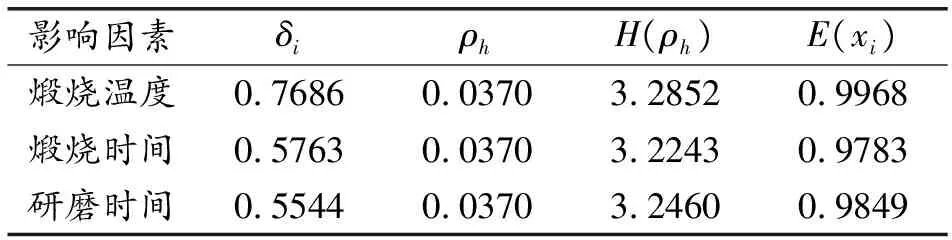
表5 灰熵关联分析相关参数计算结果
2.4 机理分析
根据上述分析结果可知,600 ℃煅烧2 h,研磨2 h所得青稞秸秆灰的活性最高。为了进一步分析其活性效应产生的原因,对其微观结构进行测试,FTIR及XRD的测试结果如图7所示。由FTIR图可以看出,HBSA的FTIR谱图主要由5个特征吸收谱带组成。其中,位于464 cm-1处的吸收谱带是由Si-O-Si键的对称变角振动引起的,位于800和1 048 cm-1处的吸收谱带是由Si-O-Si的对称和反对称伸缩振动引起的。这些特征谱带的存在,说明了HBSA中主要成分为SiO2。位于1 609和3 435 cm-1处的吸收谱带是分别由结晶水的变角振动和伸缩振动引起,说明了HBSA中含结晶水化合物的存在。由XRD图谱可以看出,HBSA最主要的衍射峰为SiO2,还有部分CaAl2Si2O8·4H2O存在。HBSA的矿物学成分主要为石英和斜方钙沸石。此外,HBSA的XRD图谱表现出较强的非晶态性质,说明HBSA中的SiO2大部分是以无定型形态存在的。
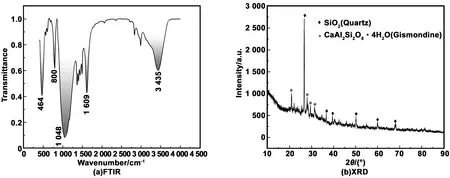
图7 HBSA的FTIR和XRD图谱
通过XRF测试得到煅烧温度为600 ℃、煅烧时间为2 h、研磨时间为2 h所得HBSA的化学组成,见表6。HBSA的主要成分为SiO2,含量达到61.751%。SO3的含量为1.75%,满足活性材料要求不超过3.5%的规定[20]。通过实验测得HBSA的烧失量(LOI)为4.55%,满足活性材料要求的不超过10%的规定[20]。可见,HBSA的各项指标均较好地满足火山灰材料的性能要求。因此,HBSA是一种适用于水泥基材料的活性掺合料。根据前述分析可知,600 ℃煅烧2 h所得HBSA的结晶度为21.54%,因此,活性SiO2的含量达到全部化学成分的48.45%。较高的活性SiO2含量,构成HBSA作为活性混合材料的必要条件。

表6 HBSA的化学组成
掺入最佳制备条件HBSA的对比试件以及未掺HBSA的MOCM标准试件进行SEM测试的微观形貌,如图8所示。显然,未掺HBSA的MOCM中产物基本以针束状的5相晶体为主,未明显发现水化硅酸镁(M-S-H)凝胶的存在,且结构较为疏松,有大量的孔洞存在。而掺入HBSA的MOCM中,明显有大量的M-S-H凝胶形成,且均匀的包裹在5相晶体表面,形成凝胶-晶体共聚物。M-S-H凝胶是一种致密的层状硅酸盐结构,主要包括滑石、海泡石和蛇纹石结构[21]。此外,由于M-S-H凝胶的形成,大量的孔洞被有效填充,结构变得更加密实。基本的水化反应过程如下:

图8 MOCM的SEM图
第一水化阶段:
5MgO+MgCl2·6H2O+7H2O→5Mg(OH)2·MgCl2·8H2O
MgO+H2O→Mg(OH)2
第二水化阶段:
3Mg(OH)2+2SiO2→Mg3Si2O5(OH)4+H2O(蛇纹石)
3Mg(OH)2+4SiO2→Mg3Si4O10(OH)2+2H2O(滑石)
4Mg(OH)2+6SiO2+3H2O→Mg4Si6O15(OH2)2-(OH)2·4H2O(海泡石)
综合上述活性指数测试结果及微观结构分析,一定条件下煅烧及研磨所得HBSA具有较高的活性效应。其活性产生的根本原因在于:HBSA中具有较高的SiO2含量,且大部分SiO2以非晶态形式存在。活性SiO2能够与氯氧镁水泥的水化产物Mg(OH)2发生二次水化反应,生成M-S-H凝胶。HBSA掺入MOCM中,将其不利产物Mg(OH)2转为有利产物M-S-H凝胶。M-S-H凝胶可以有效地填充固体颗粒间隙,使得结构更加致密,提高了力学性能。
3 结 论
通过微观及宏观测试手段对HBSA的活性性能进行了分析,并对影响HBSA活性的因素以及产生活性的机理进行了分析,得出以下结论:
(1)600 ℃二次煅烧2 h所得HBSA的XRD结晶度最小,研磨2 h所得HBSA的平均粒径最小、比表面积最大、活性最高。该条件下制备的HBSA掺入MOCM中,相对活性指数最大。
(2)基于灰熵关联分析结果,煅烧温度、煅烧时间、研磨时间三个因素对HBSA的活性效应影响的灰熵关联度大小依次为:煅烧温度、研磨时间、煅烧时间。煅烧温度应为活性HBSA制备过程中的首要控制要素。
(3)HBSA中含有较高的SiO2含量,且大部分SiO2以非晶态形式存在。活性SiO2与MOCM的水化产物发生二次水化反应,生成M-S-H凝胶,附着在5相晶体表面以及填充固体颗粒间隙,使得结构更加致密,力学性能提高。
