一种极性共聚合固定相的制备及其对碱性物质的色谱分离研究
2022-01-13余婷玉高运苓
余婷玉, 蔡 爽, 高运苓, 李 静*,3
(1.湖北文理学院附属医院,襄阳市中心医院中心实验室,湖北襄阳 441021;2.湖北文理学院食品科学技术学院·化学工程学院,湖北襄阳 441053;3.湖北文理学院附属医院,襄阳市中心医院,白癜风特色制剂襄阳市重点实验室,湖北襄阳 441021)
反相色谱(Reversed-Phase Liquid Chromatography,RPLC)的分离效率高、分析对象覆盖范围广,且可与多种检测器兼容,在日趋多样化的色谱分离模式中仍占据主流地位。其中,疏水性长链烷基键合硅胶是目前应用最广泛的反相色谱固定相[1,2]。硅胶基质具有机械强度高、化学性质稳定、粒径及孔径可控、易于表面化学修饰等诸多优势,但也存在一定的缺陷:(1)在进行键合相修饰前,通常会采用酸或热处理对硅胶进行再次羟基化以增加硅羟基的数量、提高功能基的键合率[3]。然而,约有50%的硅羟基由于空间位阻无法参与键合反应,以形成氢键的结合状态或游离状态残留,其中游离的硅羟基呈现较强的酸性,在色谱分析中易与碱性化合物以及富含-COOH、-NH2等亲水性基团的多肽或蛋白质等大分子物质产生较强的相互作用,造成峰拖尾甚至发生不可逆吸附,大大降低分离度与灵敏度[4];(2)长链烷基键合硅胶在高比例水相的流动相中易出现疏水塌陷,RPLC中最常用的C18色谱柱尤为明显,因而对于极性物质的分离能力十分有限[5]。
为改善上述缺陷,色谱学者们提出了极性修饰的反相色谱固定相[6]。在硅胶与疏水烷基长链之间插入酰胺基、脲基、磺酰胺基等极性基团,称为极性嵌入,合成过程通常需要分两步进行,但由于空间位阻影响,第二步反应产率有限,最终得到的固定相表面基团不均匀,且残余的活性极性基团会降低烷基的疏水分离性能;以含氨基、氯、羟基等极性基团的短链硅烷化试剂代替传统的三甲基氯硅烷等封端剂对硅胶基质中的残留硅羟基进行封闭称为极性封端,这种方法得到的固定相具有与普通的烷基键合固定相相似的反相保留特性,同时可改善长链烷基键合硅胶在高比例水相中的疏水塌陷问题,但与极性嵌入相比,极性基团的负载量较低,因此掩蔽硅羟基的效果稍差[6 - 8]。
1993年,Wirth等提出通过“水平聚合”的方式在硅胶微球表面同时键合长链C18和短链C3:先将干燥的硅胶置于湿润氮气中,使其吸附一层水分子,而后继续在无水环境下进行键合。在单分子水层的作用下,十八烷基三氯硅烷和丙基三氯硅烷在硅胶表面水解并通过Si-O-Si产生水平方向聚合[9]。他们在后续研究中还发现,通过水平聚合法制备的键合硅胶C18负载密度更高,残余硅羟基的数量也得以减少;且Si-O-Si在硅胶表面的密集分布形成了一层膜状的“空间屏障”,使其能够耐受极端pH值的流动相[10,11]。梁鑫淼课题组在水平聚合的基础上提出了一种改良极性封尾方法,以含有极性基团的短链烷基代替C3,得到同时具有疏水长链烷基和极性短链的键合硅胶,称为“极性共聚合(Polar-copolymerization)”[12],并将该合成方法引入到多种类型固定相的合成中,实现了生物碱和蛋白质的良好分离[13 - 18]。由于水平聚合能够有效提高硅烷键合率,因此通过极性共聚合得到的新型固定相碳负载量更高、稳定性更强、极性封尾效果更好。
在本研究中,我们通过极性共聚合法(PC法)对硅胶进行C18和二醇基共修饰,在不同条件下制备一系列极性共聚合固定相(PCSP),详细考察在极性共聚合过程中,硅胶润湿度、极性硅烷偶联剂投料量等反应条件对硅羟基掩蔽效果的影响;同时与采用传统键合法(Conventional Bonding,CB法)制备的固定相(CBSP)进行比较,深入探讨了PCSP在碱性物质分离分析中的多重优势。
1 实验部分
1.1 仪器及试剂
JSM-35CF扫描电镜(日本,JEOL);TriStarⅡ全自动比表面分析仪(美国,Micromeritics);AVATAR 360傅立叶变换红外光谱仪(美国,Thermo);TG 209 F1 Libra热重分析仪(德国,NETZSCH);LC-20A高效液相色谱仪(日本,Shimadzu);DDS -307电导率仪(上海精密科学仪器)。
硅胶基质来源于实验室自制(合成方法参照前期工作[19],粒度5~7.5 μm);阿米替林、菲、秋水仙碱、2-萘胺、延胡索乙素、利多卡因,均购于阿拉丁试剂(上海)有限公司;分析纯H2SO4、HCl、甲苯购于国药集团化学试剂有限公司;十八烷基三氯硅烷(ODS)购于百灵威科技有限公司;3-缩水甘油醚氧基丙基三甲氧基硅烷(GOPTS)购于武汉大学有机硅新材料有限公司;色谱纯甲醇和乙腈购于美国Sigma-Aldrich公司。实验用水为Heal Fore NW系统(上海)制备的超纯水。
1.2 实验方法
1.2.1 活化硅胶按照1 g∶10 mL的比例将硅胶加至6 mol·L-1的HCl中,120 ℃加热回流12 h,然后用超纯水洗至中性,160 ℃真空干燥,即得到不含吸附水的干燥硅胶。
1.2.2 润湿硅胶参照文献方法[9],将干燥氮气以恒定的流速通过装有200 mL超纯水的三颈烧瓶中,烧瓶置于恒温水浴中加热,三颈烧瓶出口端连接至装有干燥硅胶的玻璃瓶,玻璃瓶出口插入湿度计,调节水浴温度使湿度维持在50%左右,经过一段时间后即得到单分子水层润湿的硅胶。
硅胶的润湿度以干燥硅胶质量增重率(+w%)计算:+w%=(m3-m2)/(m2-m1),其中m1为空瓶质量,m2为加入干燥硅胶后总瓶重,m3为经过一段时间润湿后的总瓶重。干燥硅胶的润湿度计为0%。
1.2.3 硅胶键合与装柱在三颈烧瓶中加入干燥(CB法)或润湿(PC法)硅胶,按照1 g∶10 mL的比例加入无水甲苯,在干燥氮气氛围下,加入一定比例的ODS和GOPTS,搅拌下110 ℃加热回流8 h;反应完成后所得产物依次用甲苯和甲醇抽洗3次,80 ℃干燥后加入甲醇-稀H2SO4(0.5 mmol·L-1)的混合溶液(30∶70,V/V)中,100 ℃回流3 h,水洗至中性,80 ℃干燥,即得到CBSP或PCSP;将所得固定相用甲醇匀浆,在7 000 psi下分别填装至不锈钢空柱管(100 mm×2.1 mm i.d.)中。
1.2.4 酸性硅羟基的测定与评估采用电导滴定法测定材料中酸性硅羟基的数量[4]。称取50 mg固定相材料,加入60%甲醇溶液10 mL使其均匀分散,再加入1 mL 0.1 mol·L-1HCl,然后用0.1 mol·L-1NaOH溶液进行滴定,记录体系中电导率K的变化,绘制电导滴定曲线,并根据电导率上升第一阶段消耗的NaOH溶液体积来计算酸性硅羟基的相对含量。
根据色谱柱评价参考标准文件870[20],以阿米替林为探针,根据其在各固定相上的保留时间和拖尾因子评估各固定相中的残余酸性硅羟基活性。阿米替林母液用甲醇配制成1 mg·mL-1,进样前用流动相稀释至25 μg·mL-1。色谱条件:80%甲醇-20%磷酸盐缓冲溶液(PBS,5 mmol·L-1,pH为7.0或7.6);流速:0.15 mL·min-1;柱温:25 ℃;进样量:2 μL;紫外检测波长:254 nm。
2 结果与讨论
2.1 材料合成与表征
表1列举了采用PC或CB两种方法,在不同条件下合成得到的一系列固定相及对应的热重失重率(各批次硅胶的初始投料量均为4 g)。

表1 不同条件下合成的PCSP及CBSP
图1(a)、1(b)分别为自制硅胶微球的扫描电镜(SEM)图及氮吸附曲线,自制硅胶的比表面积约为163 m2·g-1,平均孔径约为8.2 nm,累积孔容为1.15 cm3·g-1;图1(c)为P-4制备过程中各步产物的傅立叶红外光谱表征结果,当硅胶与ODS和GOPTS发生反应后,在921 cm-1出现的弱峰为环氧基C-O键的振动信号,2 860 cm-1和2 930 cm-1处的双峰为C18长链的C-H对称和不对称伸缩振动信号,在酸性环境下开环后得到P-4,环氧基转化为二醇基,921 cm-1处的特征信号几乎消失,C18特征信号无明显变化,由此表明P-4材料表面键合基团为C18和二醇基;将相同质量的C-1、P-1、P-4、P-7加入至相同体积纯水中,强烈振摇后如图1(d)所示,C-1具有极强的疏水性,漂浮于液面上且大量沾壁,P-1、P-4、P-7三种材料在水中呈现良好分散,且P-7在水中的分散性最佳,无沾壁现象,说明通过极性共聚合法掺入二醇基后可改善疏水长链烷基键合硅胶在水中的浸润性,且材料的亲水性随GOPTS投料比例的提高而增强,对于极性化合物在高比例水相中的分离十分有利。
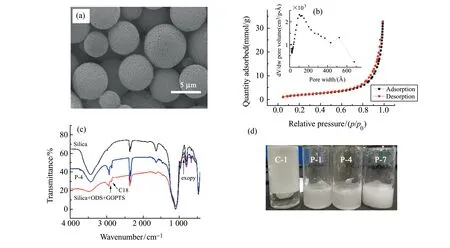
图1 PCSP及CBSP的表征(a:自制硅胶的扫描电镜(SEM)图;b:自制硅胶的氮吸附曲线及孔径分布图;c:P-4及中间体的傅立叶红外(FT-IR)光谱图;d:各固定相在水中的分散性)
2.2 硅胶润湿度对硅烷键合率的影响
P-1~P-5为在硅烷投料比相同、硅胶润湿度依次增加的条件下得到的系列PCSP,从表1可以看出:(1)PCSP的TG%高出CBSP约6%:在PCSP中,表面吸附单层水分子为ODS和GOPTS的水解带来更多的活性位点,在硅胶表面以水平方向发生聚合,形成一层Si-O-Si的“膜结构”,有效地提高了硅烷键合率;(2)硅烷键合量与硅胶润湿度呈正相关:随着+w%(0.66%~5.27%)的增加(P-1~P-4),TG%持续增加,说明在单分子水层吸附达到饱和前,吸附的水分子越多,越有利于极性共聚合的进行,但当+w%继续提高至6.97%(P-5),TG%不再有明显增加,可能是聚合反应达到最大限度,因此硅胶润湿度约5%为最佳聚合条件。值得注意的是,不同硅胶吸附水分子的能力不同,因而最佳润湿度也不一致,对于不同来源的硅胶应单独优化[21]。
2.3 硅胶润湿度对酸性硅羟基的影响
在色谱柱评价参考标准文件870[20]中,阿米替林(pKa=9.4)的拖尾因子与其保留时间关联不大,因而作为评估色谱柱峰形对称性的探针分析物。阿米替林在以C-1、P-3、P-4为固定相的色谱柱上的保留及峰形如图2所示。在C-1中,残余酸性硅羟基与碱性阿米替林发生不可逆吸附,阿米替林未能被洗脱;而在P-3和P-4中,极性基团二醇基的加入有效降低了酸性硅羟基的数量,并在一定程度上有空间屏蔽效果,因而可以削弱阿米替林与硅胶基质之间的相互作用,有效改善其拖尾或不可逆吸附现象;由于硅胶的润湿度更高,P-4键合的C18和二醇基均更多,因此阿米替林的保留时间大幅度增加,接近于P-3的2倍,拖尾因子也相比P-3进一步减小。总的来说,在反应未达到饱和的情况下,极性共聚合法对酸性硅羟基的屏蔽效果、对硅烷键合率的提高程度均与硅胶润湿度呈正相关。
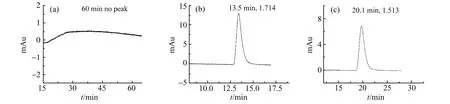
图2 阿米替林在由C-1(a)、P-3(b)和P-4(c)装填的色谱柱上的色谱图(图标为保留时间和拖尾因子)
2.4 二醇基相对含量对酸性硅羟基的影响
P-6、P-4、P-7为在相同硅胶润湿度、ODS:GOPTS的投料比逐渐增加的条件下合成得到的PCSP(表1),图3展示了阿米替林在该系列PCSP上的保留行为。鉴于当流动相pH=7.0时,阿米替林在C-1上发生不可逆吸附,因此将流动相的pH值调整为7.6,以抑制阿米替林吸附在P-6(不含二醇基、C18键合量相对C-1更高)上的吸附。在图3中,阿米替林的保留时间及拖尾因子均按照P-6、P-4、P-7的顺序依次减小,与投料中GOPTS的比例呈负相关。这是由于反应过程中,投入硅烷总量不变,ODS与GOPTS竞争键合位点,当GOPTS∶ODS的投料比逐渐增加时,制备的固定相中C18的键合量相对减少、二醇基的键合量相对增加,对于酸性硅羟基的屏蔽效果理论越强,因此阿米替林的峰形拖尾情况改善效果越好,保留时间也逐渐缩短。
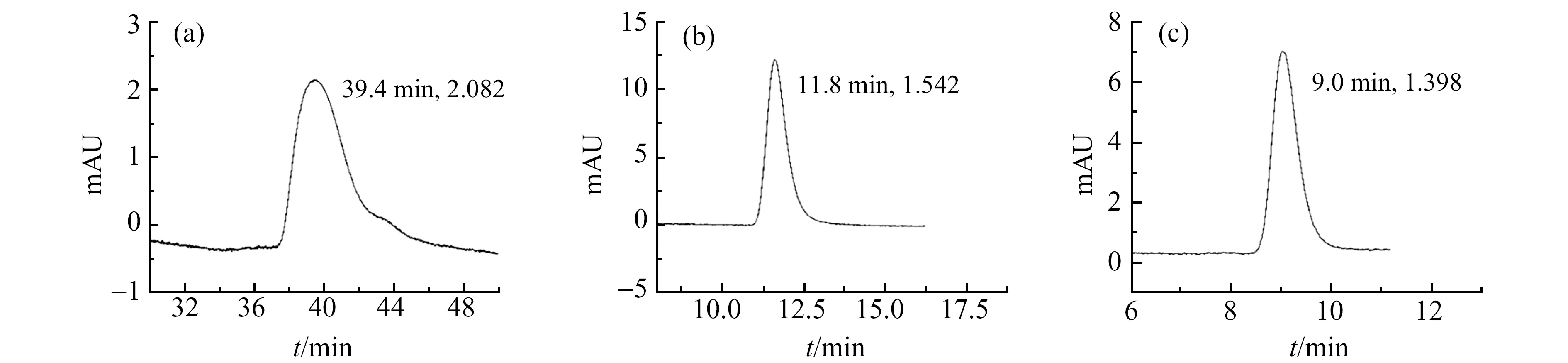
图3 阿米替林在由P-6(a)、P-4(b)和P-7(c)填装的色谱柱上的色谱图(图标依次为保留时间和拖尾因子)
通过电导滴定法测定P-6、P-4和P-7中酸性硅羟基的数量来验证上述结果,P-6、P-4和P-7在溶液电导率上升第一阶段消耗的NaOH溶液体积分别为330、200和160 μL,由此计算出各材料中酸性硅羟基的含量依次为6.6 μmol·g-1、4.0 μmol·g-1和3.2 μmol·g-1,与图3中阿米替林的出峰情况完全一致;P-6由于未加入GOPTS,酸性硅羟基数量最多,加入一定比例GOPTS后,固定相中的酸性硅羟基被二醇基短链所覆盖,P-4和P-7的含量依次降低至60%、48%,由此可证明二醇基对酸性硅羟基的掩蔽作用,且掩蔽效果与其相对键合量呈正相关。
2.5 碱性样品在PCSP与CBSP上的分离
4种代表性的小分子碱性化合物在P-4和C-1上的峰形如图4所示。秋水仙碱、2-萘胺、延胡索乙素、利多卡因依次被洗脱,在C-1上的拖尾因子分别为:1.491、1.250、2.487、2.143,在P-4上的拖尾因子分别为:1.168、0.993、1.224、1.462,均优于各自在C-1上的表现。由此可以看出,将PCSP应用于碱性样品分离时,可有效屏蔽酸性硅羟基,抑制碱性分析物的拖尾。

图4 碱性样品在C-1和P-4为固定相的色谱柱上的色谱图
3 结 论
本文通过极性共聚合法,成功制备了一系列由疏水性C18和极性二醇基共同修饰的硅胶固定相,并详细考察了硅胶润湿度、极性基团加入比例等反应条件对硅羟基掩蔽效果的影响。相比于传统方法,极性共聚合法可有效提高硅烷键合率,改善烷基键合硅胶的亲水性,有效屏蔽酸性硅羟基、改善碱性物质的拖尾;此外,极性共聚合固定相对酸性硅羟基的屏蔽效果与硅胶润湿度、二醇基的相对键合比例均呈正相关;代表性小分子碱性药物的良好分离及对称峰形显示出PCSP在碱性物质色谱分析中的应用价值。
