Challenges and suggestions for precise diagnosis and treatment of Wilson’s disease
2022-01-12DongZhiYingWu
i Dong · Zhi-Ying Wu , Y
Hepatolenticular degeneration, also known as Wilson′s disease (WD), is an autosomal recessive disorder of copper dysfunction. The causative geneATP7Bencodes a copper transporting P-type ATPase, and the pathogenic mutations withinATP7Bmay lead to impaired copper excretion via the biliary tract and to massive copper accumulation in the liver, brain, kidney, bone and joints, cornea and other tissues and organs [ 1- 3]. Therefore, individuals with Wilson’s disease may suffer liver damage, neuropsychiatric expressions, renal damage and osteoarthropathy. In general, the clinical manifestations of WD are complex and diverse, and early diagnosis is difficult. However, WD is currently one of the few treatable neurogenetic and metabolic diseases. With standardized treatment, the prognosis of patients is good and is no different than that of ordinary people. Therefore,early diagnosis and standardized treatment of WD is vital.Currently, guidelines on the diagnosis and treatment of WD have been issued successively at home and abroad [ 4- 6].Recently, we released the “Chinese guidelines for diagnosis and treatment of Wilson’s disease 2021” based on our progress and experience in recent years [ 7]. Here, we summarize the difficulties and challenges in the clinical diagnosis and treatment of WD for the benefit of improving the level of diagnosis and treatment of WD.
Difficulties and challenges of clinical diagnosis
Diagnosing typical WD patients is not difficult when using the latest version of the guidelines [ 4- 7]. However, some patients present atypical clinical manifestations. For example, commonly used clinical indicators during clinical practice, including serum ceruloplasmin, 24-hour urine copper,and corneal Kayser-Fleischer (K-F) ring, may differ from the classical manifestations of WD. Performance also can be contradictory, leading to delayed diagnosis. Clinicians should be aware of the variability and complexity of the results of these indicators, especially for the early diagnosis of WD patients in China.
Variability of serum ceruloplasmin levels
In the normal population, the level of ceruloplasmin generally ranges from 200 to 500 mg/L, whereas the level in WD patients is usually less than 200 mg/L [ 8]. In clinical practice, ceruloplasmin levels also could be reduced if with chronic liver disease, certain wasting diseases,and even other rare genetic diseases. Mutation carriers ofATP7Bgene may show low ceruloplasmin but generally not less than 50% of the lower limit of the normal reference value. Previous studies showed that ceruloplasmin level less than 80 mg/L is a strong evidence for clinical diagnosis of WD patients [ 9]. Previous analysis from large samples found that 97.5% of WD patients with a clear genetic diagnosis had an average ceruloplasmin level below 120 mg/L [ 10], indicating that this threshold provides guidance for clinical WD diagnosis. In addition,it should be noted by clinicians that a few WD patients can have normal ceruloplasmin levels, especially in the acute liver disease stage, or during other inflammatory periods or during pregnancy [ 11]. Therefore, for a patient with clinically suspected WD, other indicators need to be evaluated to avoid a missed diagnosis if the ceruloplasmin level is normal.
24-hour urine copper level for diagnosis
It is necessary to standardize the collection method of 24-hour urine copper. The urine needs to be fully mixed during testing to avoid testing errors. If 24-hour urine copper level is higher than 100 μg and with normal creatinine clearance rate, WD should be suspected and further confirmed.If children with unknown liver function abnormalities having urine copper level higher than 40 μg [ 10, 12], clinicians should be very cautious and provideATP7Bgenetic screening for suspected children.
Presence or absence of corneal K-F ring
The typical corneal K-F ring can be observed via ordinary light, which is a yellow-green or yellow-gray pigment ring on the edge of the cornea. If the K-F ring is atypical, it is necessary to observe via a slit lamp to determine whether or not. A negative K-F ring cannot be used to rule out a WD diagnosis completely. Some patients without neurological manifestations or pre-symptomatic patients may have no K-F ring. In addition, although K-F ring is a typical clinical hallmark of WD patients, it also may appear in patients with long-lasting cholestasis or cryptogenic cirrhosis [ 8], which should be recognized clinically.
Necessity of ATP7B gene screening
At present, with widespread access of genetic screening technology and the reduction of costs,ATP7Bgenetic testing can be recommended to be performed to confirm the diagnosis in patients suspected with WD clinically. Therefore, the previously recommended liver copper measurement and biopsy pathology are currently eliminated owing to their invasiveness and poor guidance value.
Our previous study found that some WD patients can simultaneously carry three mutations [ 13], suggesting that one chromosome may harbor two or more pathogenic mutations. Therefore, duringATP7Bgene screening, the affected individual should undergo co-segregation and mutation pathogenicity analysis to ensure thatATP7Bpathogenic variants identified are intransto avoid over-diagnosis and treatment. In addition, some patients may carry large-segment deletion mutations withinATP7Bgene. Therefore,when routine Sanger sequencing cannot identify pathogenic variants, it is necessary to use multiplex ligation-dependent probe amplification to large-segment deletions [ 14].
In addition, clinicians should realize thatATP7Bpathogenic variants in patients have their regional and ethical characteristics. Unlike that H1069Q is the most common mutation in Caucasian populations [ 8], the most common pathogenic variants in Chinese patients are R778L, P992L,and T935M [ 13]. Therefore, clinical priority could be given to screen for high-frequency mutations in Chinese WD patients to save time and cost of genetic testing.
Challengeas nd suggestions for the treatment
WD is one of the few neurogenetic diseases that can be treated. Appropriate regimens can be used for treatment according to the clinical manifestations of patients (Fig. 1).There are still many problems to be highlighted during the treatment. In particular, the characteristics ofATP7Bpathogenic variants in Chinese WD patients require individualized treatment. The core of WD management is to reduce copper intake and to promote copper elimination. Therefore, a lowcopper diet should be used throughout the patient's entire treatment process.
Problems when treating WD with D-penicillamine and countermeasures
D-penicillamine is the first-line drug for the treatment of WD patients in China. Owing to its low cost and the convenience of purchase channels, it is still the most commonly used decoppering agent. However, D-penicillamine can cause irreversible aggravation of clinical symptoms in WD patients[ 15]. Therefore, there are certain principles to be followed when using D-penicillamine [ 7]. Firstly, the penicillin skin test must be carried out before the drug is used, and it can be used only if the skin test is negative. Secondly, the drug dosage should be adjusted slowly. The current recommended maximum dosage is 1500 mg/day. It is generally recommended to administer them 1 hour prior to meals. During the treatment with D-penicillamine, vitamin B6 need to be supplemented to avoid vitamin B6 deficiency. D-penicillamine plus zinc and vitamin B6 should be administered at widely spaced intervals. If the symptoms get worse, the drug should be discontinued immediately. Lastly, if the patient presents with torsion cramps and severe dysarthria, it is not recommended to use D-penicillamine to avoid irreversible neurological worsening.
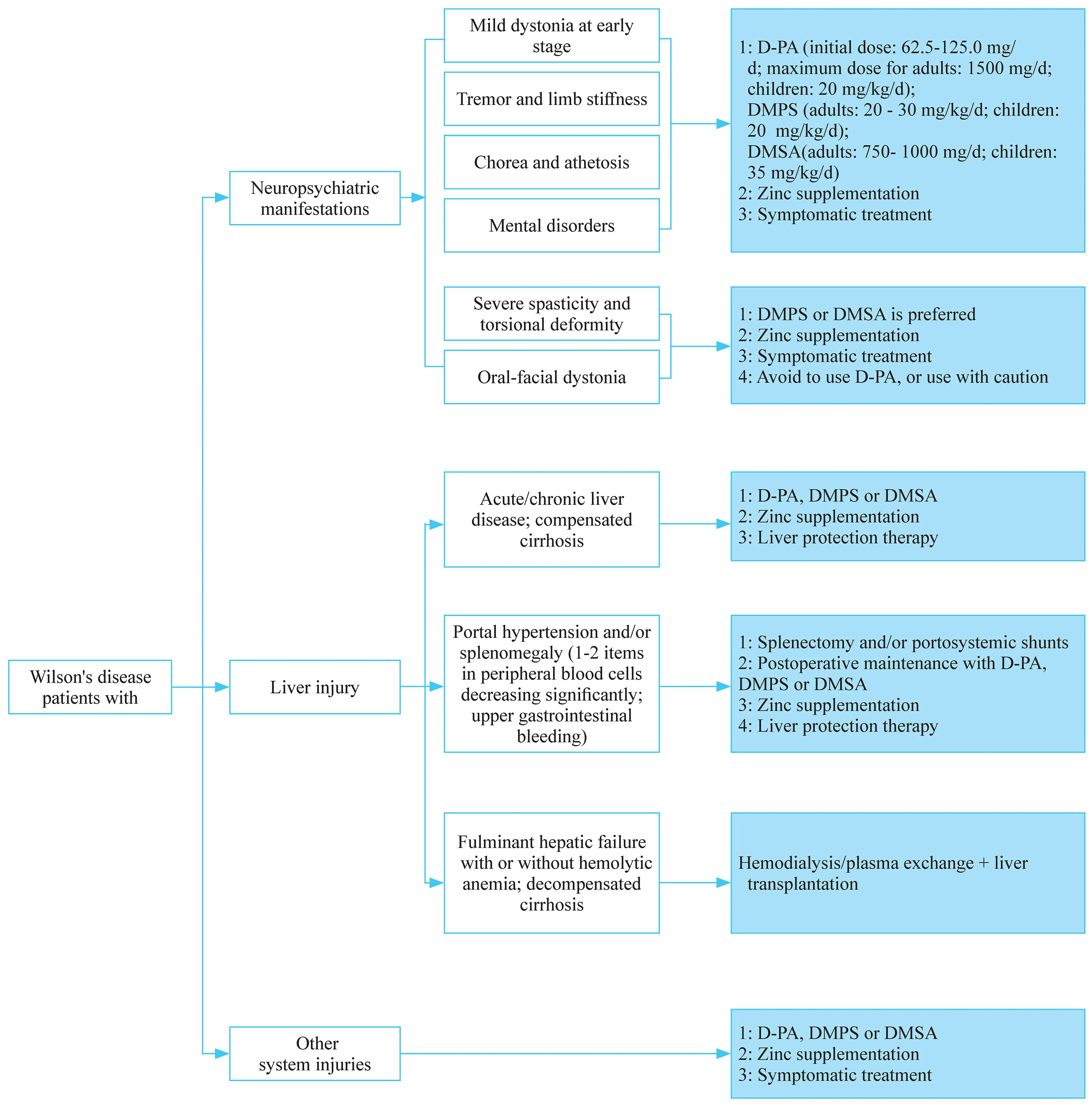
Fig. 1 Flowchart of treatment strategy in patients with Wilson's disease. D-PA D-penicillamine; DMPS sodium dimercaptopropane sulfonate;DMSA dimercaptosucinic acid
Using dimercaptosuccinic acid capsules or sodium dimercaptosuccinate: China’s experience
These agents contain two sulfhydryl groups serving as copper-exhausting effect, and are used mainly in China [ 7,16]. If WD patients are allergic to D-penicillamine or manifest severe extrapyramidal symptoms, dimercaptosuccinic acid capsules and sodium dimercaptosuccinate are recommended to be administered. It is worth noting that sodium dimercaptosuccinate may cause worsened neurological presentations in few WD patients. In addition, this drug’s copper-exhausting effect is weaker than D-penicillamine.Therefore, D-penicillamine still serves as the first choice for WD patients.
Optimal use of zinc salts
Zinc reagents have a definite efficacy in the treatment of WD, with very few side effects. The main disadvantage is that it usually takes 4-6 months to exert effects. Therefore,it is not the first choice for WD patients with serious illness,and it mainly serves as the maintenance treatment. However,for pre-symptomatic patients, pregnant patients and patients after liver transplantation, it has been used as the first-line therapy [ 7, 17, 18].
Implementing liver transplantation in WD patients with caution
The implementation of liver transplantation in WD patients is still controversial [ 18, 19]. Because WD is a genetic disease, liver transplantation only resolves liver problems. Moreover, liver transplantation requires longterm immunosuppressive therapy. Therefore, clinicians should strictly control the indications of liver transplantation in WD patients to avoid unnecessary over-treatment.Transplantation can be suitable for acute liver failure or for decompensated cirrhosis in WD patients [ 7, 20].However, liver transplantation is not recommended for patients with severe neuropsychiatric symptoms. In addition, liver transplantation cannot solve the fundamental ATP7B protein dysfunction in WD patients. Therefore,after liver transplantation, WD patients still need a lowcopper diet and zinc monotherapy [ 19], and even decoppering therapy.
Choice of splenectomy
Some WD patients may develop hypersplenism secondary to liver cirrhosis, leading to noted decrease in blood cells. If they are not treated in time, there is a risk of major bleeding. Moreover, owing to a severe decrease in white blood cells, the disease status can make D-penicillamine unavailable, which further aggravates the condition. Therefore, if patients have severe hypersplenism, moderate to severe splenomegaly, and reduced blood cells, especially WBC less than 3.0 × 10 9 /L and platelet count less than 60 × 10 9 /L, splenectomy should be performed actively [ 21].
In summary, WD currently serves as one of the few treatable neurogenetic diseases. WD treatment needs a comprehensive and collaborative multidisciplinary team including pediatrics, neurology, gastroenterology, psychology and genetics. Clinicians are required to have a deep understanding of the diagnosis and treatment modalities for WD. Early diagnosis and individualized treatment for WD patients can significantly improve the prognosis and can avoid misdiagnosis and mistreatment.
Author contributions DY contributed to writing the original draft;WZY contributed to writing, reviewing and editing.
Funding This study was supported by grants (81125009 and 81701126)from the National Natural Science Foundation of China.
Compliance with ethical standards
Ethical approval Not required.
Conflict of interest We declare that we have no conflict of interest.
杂志排行

World Journal of Pediatrics的其它文章
- Muscle strength and its association with cardiometabolic variables in adolescents: does the expression of muscle strength values matter?
- Vestibular function of pediatric patients with sudden sensorineural hearing loss: based on vertigo symptom and vestibular function testing
- Evaluation of a new frequency-volume chart for children with primary monosymptomatic nocturnal enuresis: a prospective, comparative study
- Consensus statement on the epidemiology, diagnosis, prevention,and management of cow's milk protein allergy in the Middle East:a modified Delphi-based study
- Treatment of pediatric mild persistent asthma with low-dose budesonide inhalation suspension vs. montelukast in China
- Haploidentical hematopoietic stem cell transplantation for pediatric patients with chronic active Epstein-Barr virus infection:a retrospective analysis of a single center