核壳型SiO2@松香基高分子高效液相制备柱的制备及其对喜树碱的分离纯化
2022-01-10易冯梅谢文博雷福厚
易冯梅,谢文博,李 浩,曾 磊,李 文,雷福厚
(广西民族大学 化学化工学院;林产化学与工程国家民委重点实验室;广西林产化学与工程重点实验室;广西林产化学与工程协同创新中心,广西 南宁 530006)
喜树碱(CPT)是从喜树中提取得到的一类天然植物抗癌药物[1-2]。喜树碱作为一种原料药具有重要的生产及研究价值,主要提取方法有碱法提取、超声波提取、索氏回流提取等。这些方法虽然能较好地分离纯化喜树碱,但也存在明显的不足,如碱性或酸性溶液萃取产生的废液会污染环境,从而带来后处理问题;此外,醇提取会产生副产物,超声波提取使得喜树碱结构破坏的机率大大增加,从而降低提取率[3-6]。因此,开发低毒、高效地分离纯化喜树碱的方法意义重大。制备液相色谱是一种从混合物中纯化目标物的高性能技术[7],具有分离效率高、产物纯度高、产率高和经济环保等优点,被广泛用于天然产物的分离纯化中[8-11]。该方法的关键是通过高负载、高分离度的制备柱来实现的[12],固定相作为色谱柱的核心,它的选择尤为重要[13-14]。球形硅胶因具有表面易于修饰、机械强度高、比表面积大等优点而被广泛用作色谱固定相[15-17],但硅胶表面的硅羟基使得其pH耐受性降低,同时还增加了碱性化合物的拖尾[18]。因此,可在硅胶表面键合聚合物来提高其重复性和耐久性。键合在硅胶表面的聚合物应具有较高的机械强度和适当的交联度,用来保持固定相的色谱性能[19-20]。松香是以松树分泌物松脂为原料,通过水蒸气蒸馏得到的一种天然树脂,具有经济、环保、可生物降解等优点,主要成分为枞酸,结构中的菲环骨架使其具有较好的分子刚性及药物相容性[21-23]。松香改性后得到的马来海松酸丙烯酸乙二醇酯,可与甲基丙烯酸共聚形成松香基高分子聚合物,该聚合物在结构上与生物碱相似,表面的羧基官能团可为生物碱结构中的氮原子提供结合位点形成酸碱配合物,同时具有优异的结构刚性和稳定性。因此,该聚合物可用于键合二氧化硅用作色谱固定相来分离生物碱。本课题组以松香为原料合成出了松香基高分子微球,以此为液相色谱固定相,成功分离了喜树碱[24-25]、紫杉醇[26]及多环芳烃[27],但仍存在微球粒径分布较宽的问题。基于前期的研究成果,本研究采用制备型高效液相色谱法,以粒径均一的烷基化硅球为载体,马来海松酸丙烯酸乙二醇酯为交联剂,甲基丙烯酸为功能单体,通过涂覆-悬浮自由基聚合法制备出核壳型SiO2@松香基高分子高效液相制备柱,并用来分离纯化喜树碱,以期制备原料来源广泛、绿色环保的色谱固定相,从而为喜树碱的分离纯化提供新思路。
1 实 验
1.1 原料、试剂与仪器
喜树(CamptothecaacuminataDecne.)果,购于广西南宁;马来海松酸丙烯酸乙二醇酯,广西梧州日成林产化工股份有限公司;硅胶,苏州纳微科技公司;石油醚(沸点60~90 ℃)、3-(异丁烯酰氧)丙基三甲氧基硅烷(γ-MPTMS)、喜树碱标准品(纯度≥98%)、偶氮二异丁腈,阿拉丁试剂公司;甲醇,色谱级,美国Fisher Scientific公司;其他试剂均为国产分析纯。
Nicolet iS10傅里叶变换红外光谱(FT-IR)仪,美国Nicolet公司;SUPRA55 Sapphire场发射扫描电子显微镜(SEM),德国卡尔蔡司公司;JEOL JEM-2100F场发射透射电子显微镜(TEM),日本JEOL公司;LC-20AR制备型液相色谱(LC)仪、LC-15C高效液相色谱(HPLC)仪,日本岛津公司;ASAP 2020M比表面积及微孔物理吸附仪,美国Micromeritics公司;TG209F1热重(TG)分析仪,德国耐驰公司。
1.2 核壳型SiO2@松香基高分子(SiO2@R)固定相的制备与表征
1.2.1固定相的制备 称取50 g粒径10 μm的球形硅胶置于三口烧瓶中,加入500 mL 10%盐酸,在80 ℃下油浴回流反应8 h,反应在氮气保护下进行。反应结束后抽滤,将硅胶洗至中性,真空干燥,得到活化硅胶。称取50 g活化硅胶置于三口烧瓶中,分别加入20 mL吡啶、500 mL无水甲苯和50 mL 3-(异丁烯酰氧)丙基三甲氧基硅烷,在80 ℃下油浴回流反应8 h。反应结束后,依次用甲醇、丙酮和正己烷润洗几次,抽滤,真空干燥,得到烷基化硅胶。称取0.56 g马来海松酸丙烯酸乙二醇酯交联剂、0.08 g 甲基丙烯酸、0.004 g偶氮二异丁腈(AIBN)和0.17 g致孔剂异辛烷溶于3 mL氯仿配成油相,称取硅胶质量8%的油相(硅胶质量为40 g)溶于300 mL氯仿配成混合溶液,将混合溶液均匀涂覆到40 g烷基化硅胶表面,并置于装有十二烷基硫酸钠(SDS)溶液的三口烧瓶中,在85和95 ℃下各冷凝回流2 h。产物经无水乙醇索氏提取24 h后过筛,真空干燥,得到核壳型SiO2@松香基高分子固定相。制备过程如图1所示。

图1 核壳型SiO2@松香基高分子固定相合成示意图Fig.1 Synthesis route of core-shell SiO2@rosin-based polymer stationary phase
1.2.2固定相的表征 将活化硅胶、烷基化硅胶与核壳型SiO2@松香基高分子固定相在80 ℃下真空干燥12 h,将干燥后的样品与KBr按质量比1 ∶200压成样品片,使用红外光谱仪在4000~500 cm-1范围下扫描32次。取少量样品置于4 mL PE管内,加入甲醇超声波处理使其分散均匀,使用毛细管将样品涂到金属片上,进行喷铂处理,使用SEM和TEM对样品进行形貌观察。将固定相在100 ℃进行脱气后使用比表面积及微孔物理吸附仪测定比表面积、平均孔径和孔体积。在N2保护下,使用热重分析仪测定固定相从30 ℃以10 ℃/min的速率升温到900 ℃的TG曲线。采用细胞计数试剂盒8(CCK8)法,按照ISO 10993—12:2007标准制备质量浓度为200 mg/L的浸提液,对核壳型SiO2@松香基高分子固定相进行细胞毒性检测。
1.3 核壳型SiO2@松香基高分子色谱柱的制备与评价
1.3.1色谱柱的制备 称取固定相130 g左右,用一定量的甲醇超声波分散均匀,使用装柱机在41 MPa 下将固定相填充于30 mm×250 mm的不锈钢色谱空柱,装柱30 min,待柱压变为零后,静置10 min,卸下色谱柱,装上柱头及筛板,得到核壳型SiO2@松香基高分子色谱柱(30 mm×250 mm,10 μm)。
1.3.2色谱柱的评价 以甲醇为流动相,将色谱柱的流速从0升高到20 mL/min,分别观察并记录液相色谱A、B泵的压力。以甲苯为溶质,甲醇为流动相,流速为30 mL/min,在室温条件下测定色谱柱的理论塔板数[28]。使用尿嘧啶与烷基苯(苯、甲苯、乙苯、丙苯、丁苯)为探针,流速为17 mL/min,进样量为0.85 mL,流动相为甲醇水溶液(体积分数40%~65%),以5%的间隔改变甲醇比例,对色谱柱的保留机制进行考察[29]。在流动相为体积分数50%的甲醇水溶液,采用紫外吸收检测器,检测波长为254 nm,流速为17 mL/min,柱温为室温的色谱条件下,将尿嘧啶、苯、甲苯、二甲苯和萘的混合溶液重复进样20次,来评价色谱柱的重复性[30]。
准备4类测试混合物Ⅰ~Ⅳ,通过分离这4类混合物对色谱柱进行Tanaka评价。混合物Ⅰ包括尿嘧啶、丁基苯、戊基苯、邻三联苯和苯并菲,以V(甲醇)∶V(水)=80 ∶20作为流动相;混合物Ⅱ包括尿嘧啶、咖啡因和苯酚,以V(甲醇)∶V(水)=30 ∶70作为流动相;混合物Ⅲ包括尿嘧啶、苯甲胺和苯酚,以V(甲醇)∶V(0.01 mol/L磷酸盐缓冲液,pH值7.6)=30 ∶70 作为流动相;混合物Ⅳ的组成与混合物Ⅲ相同,以V(甲醇)∶V(0.01 mol/L磷酸盐缓冲液,pH值2.7)=30 ∶70为流动相,以上4种混合物均在流速17 mL/min,检测波长254 nm的条件下进行测试[30]。
1.4 喜树碱粗提物的制备
称取干燥的喜树果50 g,粉碎至粒径约0.6 mm,用300 mL无水甲醇回流提取60 min,重复提取3次,将提取液过滤、合并、减压浓缩后依次用石油醚和氯仿萃取,收集氯仿层,减压浓缩得粗提物约10 g。粗提物经甲醇溶解配成质量浓度为1.44 g/L的溶液,经0.45 μm滤膜过滤,备用。
1.5 核壳型SiO2@松香基高分子色谱柱分离纯化条件
根据核壳型SiO2@松香基高分子高效液相色谱柱(4.6 mm×250 mm,10 μm)分离生物碱的最佳条件[24],使用从分析型色谱柱(分析柱)到制备型色谱柱(制备柱)常用的线性放大公式,其中上样量放大估算公式见式(1),流速放大估算公式见式(2):
V=(R2×L)/(r2×l)×v
(1)
U=(R2×L)/(r2×l)×u
(2)
式中:V—制备柱进样体积,mL;v—分析柱进样体积,mL;R—制备柱内径,mm;r—分析柱内径,mm;L—制备柱柱长,mm;l—分析柱柱长,mm;U—制备柱流速,mL/min;u—分析柱流速,mL/min。
色谱柱为核壳型SiO2@松香基高分子制备柱(30 mm×250 mm,10 μm)。在流动相中甲醇体积分数80%~100%,流速5~13 mL/min,柱温为室温,检测器为紫外吸收检测器,检测波长254 nm,上样量0.6~1.4 mL的条件下,分别考察流动相中甲醇体积分数(80%~100%)、上样量(0.6~1.4 mL)和流速(5~13 mL/min)对喜树碱分离纯化效果的影响。
1.6 高效液相色谱分析条件
采用HPLC分析检测核壳型SiO2@松香基高分子制备柱的纯化产物。色谱柱为shim-pack GISS C18(4.6 mm×250 mm,5 μm),流动相V(甲醇)∶V(水)=60 ∶40,流速1.0 mL/min,柱温25 ℃,检测波长254 nm,上样量20 μL。
1.7 加标回收率实验
精密称取喜树碱标准品0.005 1 g至100 mL容量瓶中,用甲醇定容,作为储备液。精密称取已知纯度(2.21%)的喜树果粗提物0.010 0 g至100 mL容量瓶中,加甲醇定容后,取8 mL溶液至10 mL容量瓶中,制备6份,1~3份依次加入储备液366 μL,4~6份依次加入储备液440 μL。甲醇定容,摇匀,得供试品溶液,计算加标回收率。
2 结果与讨论
2.1 核壳型SiO2@R固定相的表征
2.1.1SEM和TEM分析 将马来海松酸丙烯酸乙二醇酯涂覆在烷基化硅胶表面,发生自由基聚合后形成一层壳层,核壳型SiO2@松香基高分子固定相的形貌分析如图2所示。

活化硅胶activated silica gels:a.SEM;b.TEM;核壳型固定相core-shell stationary phase:c.SEM;d.TEM图2 不同样品的SEM和TEM图Fig.2 SEM and TEM images of different samples
由图2(a)和(c)可以看出,活化硅胶和固定相均具有良好的球形结构,粒径约10 μm,且分布较均一;与活化硅胶相比,固定相表面粗糙程度增加,存在附着物;比较图2(b)和(d)可知,固定相的表面附着物清晰可见。上述结果表明,已成功制备了核壳型SiO2@松香基高分子固定相。
2.1.2孔隙结构分析 活化硅胶与核壳型SiO2@松香基高分子固定相的比表面积和孔结构如表1所示。从表1可以看出,固定相的比表面积、平均孔径和孔体积均小于活化硅胶。这可能与松香基高分子键合到硅胶表面堵住了原有的孔结构而引起数值的减小。

表1 不同样品的孔隙结构Table 1 Pore structure of different samples


a.硅胶silica gels;b.烷基化硅胶silanated silica gels;c.核壳型固定相core-shell stationary phase
2.1.4TG分析 对核壳型SiO2@松香基高分子固定相进行热重分析,结果见图4。

a.硅胶silica gels;b.烷基化硅胶silanated silica gels;c.核壳型固定相core-shell stationary phase
由图4可知,在400 ℃左右,固定相质量损失加剧,800 ℃时固定相的质量残留率为86.8%;与烷基化硅胶相比,质量损失率增加了4.51个百分点,主要为固定相上键合的松香基高分子聚合物热分解所致。由此可进一步证明核壳型SiO2@松香基高分子固定相已成功制备,同时也说明固定相具有良好的热稳定性。
2.1.5细胞毒性检测 核壳型SiO2@松香基高分子固定相的细胞毒性检测结果见图5。由图5可知,在CCK8质量浓度为200 mg/L时,固定相的相对细胞存活率为88.72%,而商业树脂苯乙烯型树脂的相对细胞存活率为82.00%。相比商业树脂,固定相细胞毒性更低,安全环保,绿色无害,可用于喜树碱的分离纯化。
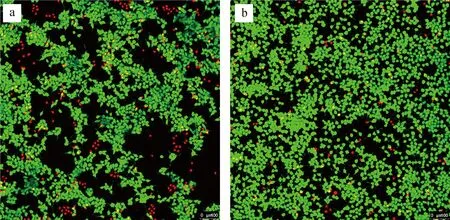
红色red:死细胞dead cells;绿色green:活细胞living cellsa.苯乙烯型树脂styrene-based anionic resin;b.核壳型固定相core-shell stationary phase图5 不同样品的细胞毒性图Fig.5 The cytotoxicity of different samples
2.2 核壳型SiO2@R制备柱的评价
2.2.1理论塔板数和流通性分析 根据1.3.2节方法测得核壳型SiO2@松香基高分子制备柱的理论塔板数为59 992。为测定制备柱的流通性,考察不同流速条件下液相色谱泵压的变化,结果见图6。由图6可知,在0~20 mL/min范围内,压力随着流速的增加而增加,呈现较好的相关性,R2值为0.989 8,结果表明制备柱具有较好的流通性。

图6 SiO2@R制备柱的流速与泵压关系Fig.6 The relationship between flow rate and pumping pressure of SiO2@R
2.2.2保留机制分析 选取苯、甲苯、乙苯、丙苯和丁苯为代表性测试物,对制备柱进行保留机制考察,结果见图7。由图7可知,保留因子反映了组分在柱中的迁移速率,其值越大,化合物在该分析条件下的保留时间越长。随着流动相中甲醇体积分数从40%增加到65%,5种疏水性测试物的保留在制备柱上呈现出逐渐减弱的趋势,符合反相色谱的保留特征。因此,核壳型SiO2@松香基高分子制备柱具有典型的反相色谱保留机制。
2.2.3重复使用性能分析 将尿嘧啶、苯、甲苯、二甲苯和萘的混合溶液重复进样20次,选取1~20次色谱峰作图,结果如图8所示。由图可知,20次的色谱图基本重合,多次进样后,此制备柱仍能达到较好的分离效果,结果表明核壳型SiO2@松香基高分子制备柱具有较好的重复使用性能。

图8 SiO2@R制备柱重复分离混合物的色谱图Fig.8 Chromatograms of repeated separation of mixtures on SiO2@R
2.2.4Tanaka评价 测试4类混合物,得到6个参数做成雷达图[30],结果见图9。
从图9可以看出,核壳型SiO2@松香基高分子制备柱的疏水性小于C18制备柱,这可能与松香基高分子聚合物中的功能基团—COOH有关;空间选择性明显大于C18制备柱,表明该制备柱适合分离结构中含有类似菲环骨架的天然药物,可能是SiO2表面聚合的松香基高分子聚合物的菲环骨架在保留机制中发挥了作用。
2.3 核壳型SiO2@R制备柱分离喜树碱
2.3.1分离条件优化
2.3.1.1最佳流速的选择 甲醇作为流动相组分具有价格低廉、易分离的特点,同时喜树碱溶于甲醇,因此选择甲醇作为流动相组分。根据线性放大之后的理论流速应为13 mL/min,根据理论流速考察不同流速(5~13 mL/min)下主成分的分离效果,其他色谱条件为流动相为95%甲醇/水、进样体积0.85 mL,结果见图10(a)。由图10(a)可知,随着流速的增加,分离时间逐渐缩短,喜树碱与其相邻杂质峰之间的分离度先升高后下降。当流速为7和9 mL/min时,分离度分别为2.942和2.899;7 mL/min时分离时间更长,更有利于其他杂质的分离。综合考虑分离度、主成分的出峰时间和经济成本,选择7 mL/min为最佳流速。
2.3.1.2流动相比例的选择 除了流速,流动相的比例也对样品的分离效果和分离时间有影响。通过优化甲醇水溶液中甲醇体积分数使样品达到理想的分离效果。在流动相流速为7 mL/min、进样体积为0.85 mL条件下,考察了流动相中不同甲醇体积分数(80%~100%)对分离效果的影响,结果见图10(b)。由图10(b)可知,随着流动相中甲醇体积分数的降低,出峰时间延迟;与此同时,喜树碱与其相邻杂质峰之间的分离度先升高后降低。当使用95%甲醇/水为流动相时分离度为3.096,达到最大,且分离时间在30 min以内,故选择甲醇/水体积比95 ∶5为流动相最佳组成比例。
2.3.1.3最佳上样量的选择 制备色谱需要在不影响纯度的情况下获得最大制备量来提高工作效率。上样量是影响样品分离度和纯度的一个重要因素。随着上样量的增加,主成分与相邻杂质间的分离度会逐渐减小,杂质峰与主成分重叠影响纯度。根据线性放大之后的理论进样体积为0.85 mL,在样品质量浓度为1.44 g/L的条件下,根据理论进样体积考察不同上样量(以换算后的进样体积0.6~1.4 mL计)对分离度的影响,结果见图10(c)。

a.流速flow rates;b.甲醇体积分数methanol volume fraction;c.进样体积sample volume图10 不同液相条件下SiO2@R制备柱分离喜树果粗提物的色谱图Fig.10 Chromatograms of crude extracts of C. acuminata fruit separated by SiO2@R at different liquid conditions
由图10(c)可知,随着进样体积的增加,喜树碱与相邻杂质峰之间的分离度逐渐减小,由0.6 mL时的1.75减小到1.4 mL时的1.48。当上样量为1.4 mL时,相邻组分无法实现分离;当进样体积为1.2 mL时,喜树碱与前后杂质峰的分离度均大于1.5。同时考虑产品的纯度及分离效率,选择进样体积为1.2 mL,经换算上样量约为1.728 mg。综上所述,使用核壳型SiO2@松香基高分子制备柱分离纯化喜树碱的最佳色谱条件如下:流动相为甲醇/水(体积比95 ∶5),流速为7 mL/min,进样体积1.2 mL(上样量约1.728 mg),检测波长为254 nm。在该条件下对喜树碱粗提物进行分离纯化,在24.65~25.41 min处收集馏分,经减压浓缩除去溶剂得到喜树碱。
2.3.2喜树碱的纯度测定 采用1.6节的HPLC方法检测,结果见图11。喜树果粗提物的纯度为42.30%,得率约20%。经核壳型SiO2@松香基高分子制备柱分离所得的喜树碱化合物的纯度为88.08%。传统硅胶不能将喜树碱与相邻杂质峰分开,所得纯化产物纯度仅为1.26%。与传统硅胶相比,核壳型SiO2@松香基高分子制备柱的纯化效果提高了86.82个百分点。与喜树果粗提物相比,经制备柱分离纯化所得喜树碱目标化合物的杂质峰明显减少,其纯度提高了45.78个百分点。
2.3.3加样回收率测定 采用1.7节的方法,计算加标回收率,平均加标回收率为96.46%,小于100%,说明固定相对喜树碱存在一定的不可逆吸附作用。RSD为1.45%,小于3%,说明回收率好,加标回收率<100%。
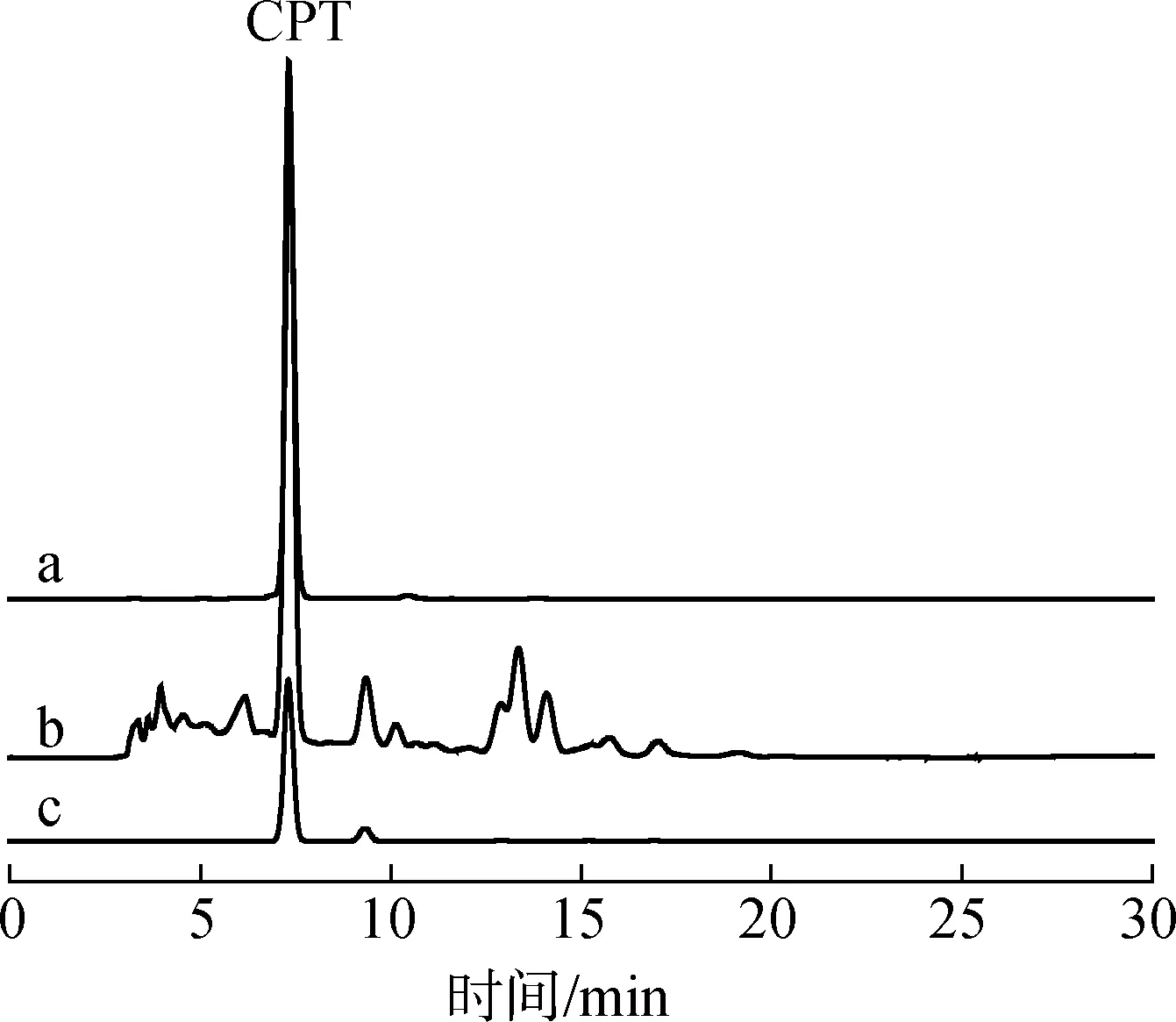
a.CPT标准品standard;b.喜树果粗提物 crude extracts from camptotheca;c.目标化合物 target compound图11 不同样品的HPLC图Fig.11 HPLC chromatograms of different samples
3 结 论
3.1以粒径均一的烷基化硅球为载体,甲基丙烯酸为功能单体、马来海松酸丙烯酸乙二醇酯为交联剂、偶氮二异丁腈为引发剂,通过涂覆-聚合法合成了核壳型SiO2@松香基高分子色谱固定相。采用红外光谱、扫描电镜、透射电镜、热重分析仪、比表面积及微孔吸附分析等方法对固定相进行了表征,结果表明:核壳型SiO2@松香基高分子固定相已制备成功,且固定相的细胞毒性低于商业树脂。
3.2采用湿法装柱制备了核壳型SiO2@松香基高分子制备柱(30 mm×250 mm,10 μm),并用于喜树果粗提物的分离纯化。实验结果表明:制备柱的理论塔板数为59 992,具有较好的流通性和重复使用性。在室温、甲醇/水(体积比95 ∶5)为流动相、检测波长254 nm、流速7 mL/min和进样体积1.2 mL(上样量约1.728 mg)的优化条件下,经分离纯化可得到纯度为88.08%的喜树碱,比浸膏初品的纯度提高了45.78个百分点。该制备柱固定相制备方法简单,原料绿色环保,方法高效、快速、可靠,操作简单,为喜树碱的分离纯化提供了新思路。
