分散固相萃取-超高效液相色谱-三重四极杆质谱法快速检测奶粉中16种喹诺酮药物残留
2021-12-31李涛周艳华向俊徐文泱孙桂芳
李涛,周艳华,向俊,徐文泱,孙桂芳
(1.湖南省产商品质量检验研究院,食品安全监测与预警湖南省重点实验室,长沙 410111;2.长沙环境保护职业技术学院,长沙 410004)
0 引言
牛奶是一种优质蛋白质脂肪来源,而液态奶营养丰富极易腐败变质,将液态奶干燥工艺处理后得到乳粉,易于长期贮存,并营养强化了维生素矿物质,如婴幼儿配方奶粉、成人奶粉等。而随着乳制品消费量逐年增加,人们特别关注乳制品安全,奶粉的主要消费群体为儿童和老人,抵抗力弱,更需要安全无药物残留的食品。喹诺酮是一类具有4-喹诺酮结构,具有抗菌谱广、活性强、价格低等特点,广泛应用于人类和动物疾病的治疗[1-2]。为获取超额经济利益,部分养殖者为逃避监管,变换喹诺酮使用类别,兽药残留的滥用超范围使用仍然存在,从而导致动物源产品中残留药物残留,低浓度的兽药残留也会产生毒性作用,如过敏、细菌耐药,也可能对人体造成不良反应[3-5]。我国《动物性食品中兽药最高残留限量》[6]规定:牛奶中恩诺沙星残留量(与环丙沙星之和)≤100μg/kg,达氟沙星残留量≤30μg/kg,氟甲喹残留量≤50μg/kg,其他沙星类如氧氟沙星、诺氟沙星、培氟沙星等均不得检出。
目前,喹诺酮化合物检测主要有HPLC法[7-8]、ELISA法[9]、LC-MS/MS法[10-11]等。液相色谱法对于少数几种喹诺酮类化合物具有良好灵敏度;而酶联免疫法前处理简单,适用于基层检测部门的初检筛查。液质联用法具有灵敏度高、选择好等优点,是兽药残留定量定性分析的首选方法。GB/T 20366-2006[12]和GB/T 21312-2007[13]是现行国家标准中动物源性食品中喹诺酮类的国家推荐标准。GB/T 21312-2007采用HLB固相萃取柱净化,前处理步骤繁杂,且没有牛奶中有限量要求的达氟沙星项目,而GB/T20366-2006未对牛奶基质进行验证研究,不适用于奶粉中喹诺酮药物残留检测。
试验拟分散固相萃取法对对奶粉中16种喹诺酮化合物留净化处理,超高效液相色谱串联三重四极杆质谱检测,并进行方法学验证和基质效应评价,建立了一种分散固相萃取-超高效液相色谱-串联质谱法快速检测奶粉中16种喹诺酮药物残留检验方法。
1 材料与方法
1.1 材料与试剂
奶粉,市售。
色谱纯乙腈、甲酸,上海CNW公司;16种喹诺酮混合标准溶液(恩诺沙星、环丙沙星、氧氟沙星、诺氟沙星、培氟沙星、洛美沙星、达氟沙星、依诺沙星、沙拉沙星、二氟沙星、司帕沙星、西诺沙星、氟甲喹、氟罗沙星、奥比沙星、麻保沙星)100μg/mL,天津迈迪嘉科技公司。
1.2 仪器与设备
TSQ Quantis型超高效液相色谱串联三重四极杆质谱(配有电喷雾离子源),美国Thermo Fisher公司;BSA224s型电子分析天平,赛多利斯科学仪器(北京)有限公司;TG16-WS型高速离心机,长沙市湘仪仪器有限公司;KQ-500DE超声波清洗器,昆山美美超声仪器有限公司;945066型数显型多管旋涡混合器,美国Talboys公司。
1.3 液相色谱条件及质谱条件优化
液相色谱条件为:A:0.1%(v/v)甲酸-5 mmol/L乙酸铵溶液,B:乙腈;流速0.25 mL/min;色谱柱温30℃;进样体积5μL,采用梯度洗脱模式。梯度洗脱程序见表1。在HESI离子源下,毛细管电压3 500 V;雾化器温度350℃,离子传输管温度350℃;鞘气压4.58 L/min;辅助气压7.97 L/min,反吹气1.5 L/min;取标准溶液直接进质谱,通过SIM和SRM扫描,确定最优质谱条件。

表1 梯度洗脱程序
1.4 样品前处理
精密称取乳粉1.0 g,加入4 mL纯水,振摇3 min后超声15 min,加入16 mL 1%甲酸乙腈,振荡15 min,离心,取4.0 mL上清液,加入分散固相萃取剂,振荡10 min后离心,上清液氮吹至近干,10%乙腈水溶液定容至1.0 m L,过0.22μm有机滤膜,供超高效液相质谱串联质谱仪测定。
1.5 方法学验证
取空白乳粉,配制2.0~100 ng/m L基质线性系列溶液,上机测定,绘制基质标准曲线。在空白乳粉添加低浓度混合标准溶液,按最优的前处理工艺处理后测定,S/N≥3的浓度为检出限(LOD),S/N≥10的浓度为定量限(LOQ)。分别在空白乳粉中加入16种喹诺酮类混合标准溶液,加标后样品浓度分别为2.0,4.0,20.0μg/kg(n=6)。测定16种喹诺酮药物残留的含量,计算回收率和相对标准偏差值。
1.6 基质效应评价
取空白乳粉,配制基质标准系列溶液,同时配制相同浓度的溶剂标准系列溶液,经液质联用仪测定后得基质标准曲线和溶剂标准曲线。依据ME=(基质标准曲线斜率/溶剂标准曲线斜率)×100评价16种喹诺酮化合物的基质效应[14]。
1.7 样品检测
取20批次乳粉,用所建立的检测方法检测样品中16种喹诺酮类化合物,判定样品中是否存在喹诺酮药物残留。
2 结果与讨论
2.1 质谱条件优化
通过标准物质的CAS号查询喹诺酮化合物分子量,通过SIM扫描,16种喹诺酮均在正离子模式下电离响应度最高,通过SRM扫描,得到喹诺酮质谱参数,以信号强度最高的离子为定量离子,信号强度较强的离子为定性离子,16种喹诺酮类质谱参数见表2。与高分辨质谱相比较[15],因其特殊的检测器,高分辨质谱在质量精度和快速定性筛查具有较大优势,而三重四极杆质谱在多化合物的高通量定量检测具有较大优势,多反应离子监测,一个母离子选一个子离子,在最优碰撞电压下,母离子碰撞产生的子离子中只选一个离子,两次都选单离子,而高分辨质谱是母离子碰撞产生多个子离子,因此三重四极杆质谱噪音干扰排除的更多,灵敏度信噪比更高,稳定性也更好。在最优质谱条件下,16种喹诺酮定量离子提取色谱图见图1,由图1可知,16种喹诺酮化合物在12 min内得到了有效分离。由于喹诺酮化合物种类较多,乙酸铵溶液可改善喹诺酮峰形,分离度更好,低浓度甲酸可为喹诺酮化合物电离时提供质子,使其响应度和灵敏度更好。
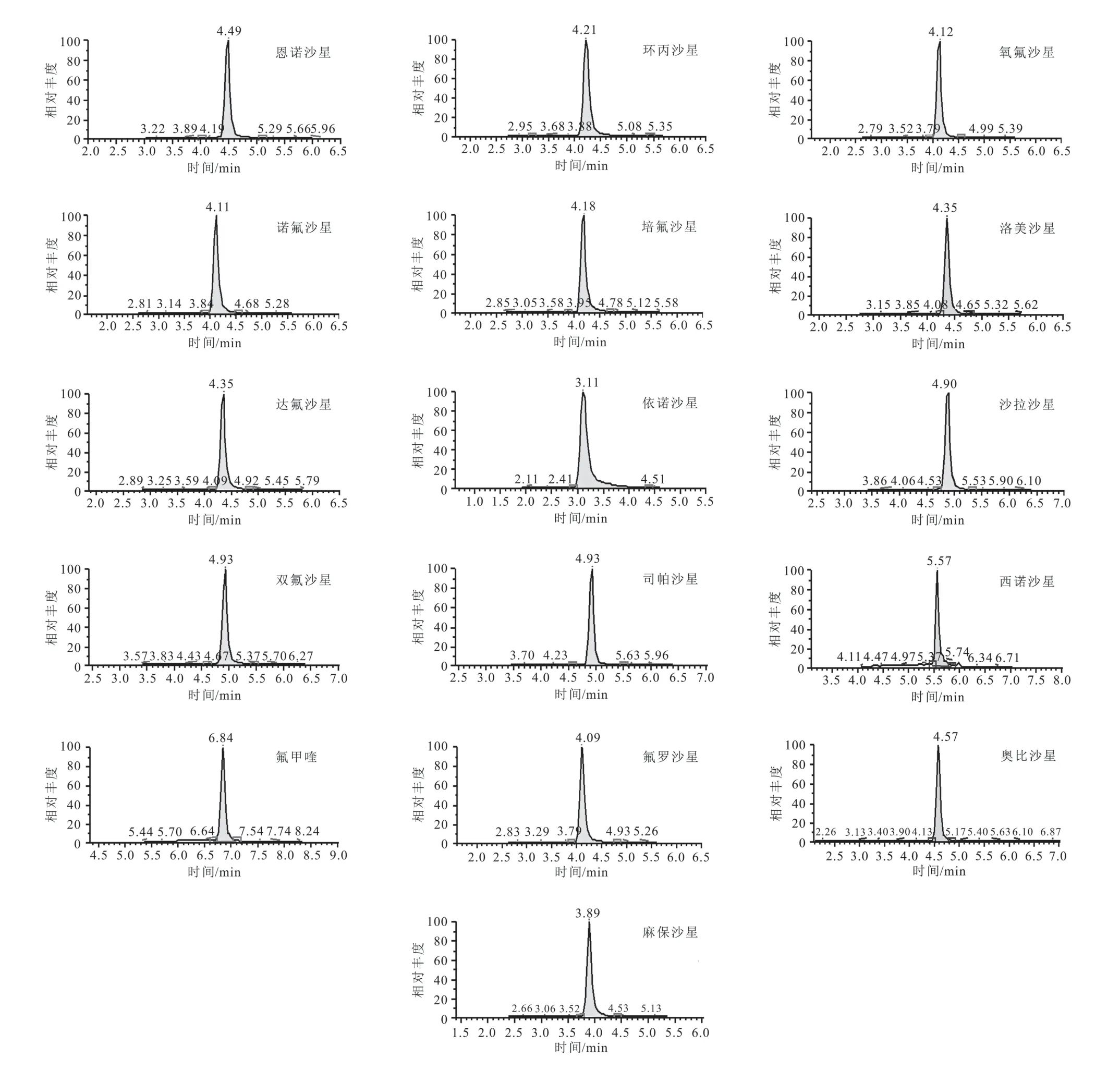
图1 喹诺酮类混合标准溶液定量离子色谱图
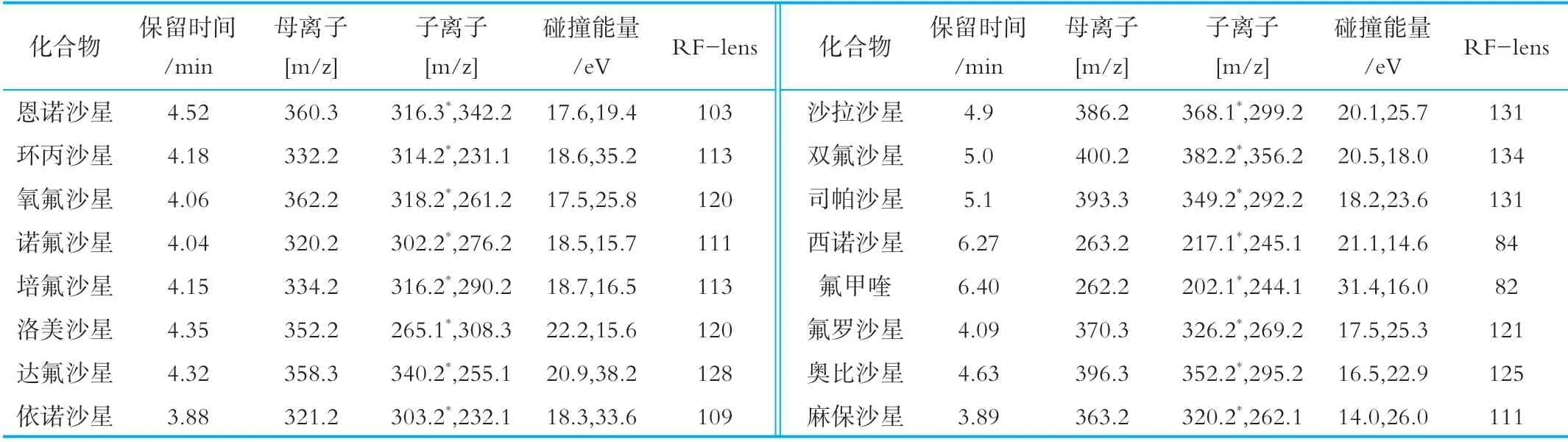
表2 16喹诺酮类化合物的质谱参数表
2.2 净化剂优化
本研究比较了C18吸附剂、PSA吸附剂、NH2吸附剂、HLB吸附剂4种吸附剂对喹诺酮净化效果。通过前处理步骤获得含有16种喹诺酮药物残留的提取液,4 m L提取液分别加入200 mg吸附剂,振荡离心过膜后测定目标化合物,计算回收率,回收率见图2。结果表明,经C18吸附剂和PSA吸附剂净化后的回收率最高,16种喹诺酮回收率均高于70%,经HLB吸附剂处理后,氧氟沙星和达氟沙星回收率较低,经NH2吸附剂处理后,9种喹诺酮类回收率低于60%。NH2是由硅胶键合氨丙基,兼具极性吸附作用和弱阴离子交换作用,可通过弱阴离子交换(水溶液)或极性吸附(非极性有机溶液)达到保留作用。HLB吸附剂是一类用于酸性、中性和碱性化合物的通用型吸附剂,对极性化合物具有较强的保留能力。PSA吸附剂是一类药物残留分析前处理中分散固相萃取的常用净化剂,PSA可与金属离子产生鳌合作用,可以有效去除脂肪酸,有机酸,和一些极性色素及糖,C18吸附剂,具有疏水作用,对非极性的组分有吸附作用,主要用于反相萃取,适合于非极性到中等极性的化合物。奶粉基质复杂,含有蛋白质、脂肪、金属离子等,为确保净化效果,本研究拟选择C18吸附剂和PSA吸附剂进行净化处理。
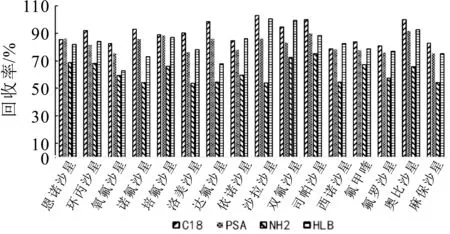
图2 不同吸附剂对喹诺酮回收率的影响
2.3 净化剂组合的优化
单一净化剂难以获得多药物残留较为满意的净化效果,本研究在单一吸附剂的基础上,选取了净化效果较好的C18吸附剂和PSA吸附剂,对C18吸附剂和PSA吸附剂组合的用量进行了优化。4 mL提取液分别加入吸附剂组合分别为100 mg C18+50、200 mg C18+100 mg PSA、300 mg C18+150 mg PSA,振荡离心过膜后测定目标化合物,计算回收率,回收率结果见图3。不同吸附剂用量对喹诺酮化合物回收率的也具有较大影响,当添加量为C18吸附剂用量为200 mg,PSA用量为100 mg时,各类化合物的回收率最高,当C18吸附剂用量为300 mg时,目标化合物的回收率反而降低,说明当吸附剂用量较高时,会对目标化合物产生吸附,从而降低了目标化合物的回收率,当吸附剂组合用量较低时,净化不完全及净化效果不好也会影响到目标化合物的回收率。

图3 不同吸附剂用量对喹诺酮回收率的影响
2.4 线性关系、检出限和定量限结果分析
由表3可知,16种喹诺酮浓度为2.0~100.0 ng/mL范围内,线性关系良好(R2≥0.997),检出限(LOD)范围为0.03~0.4μg/kg,定量限范围(LOQ)为0.1~1.5 μg/kg,此方法所得大部分喹诺酮化合物的检出限和定量限优于GB/T 20366-2006和GB/T 21312-2007。不同喹诺酮化合物的分子量和结构不同,在同一条件下的响应度和信噪比不同,因此检出限和定量限也不同。
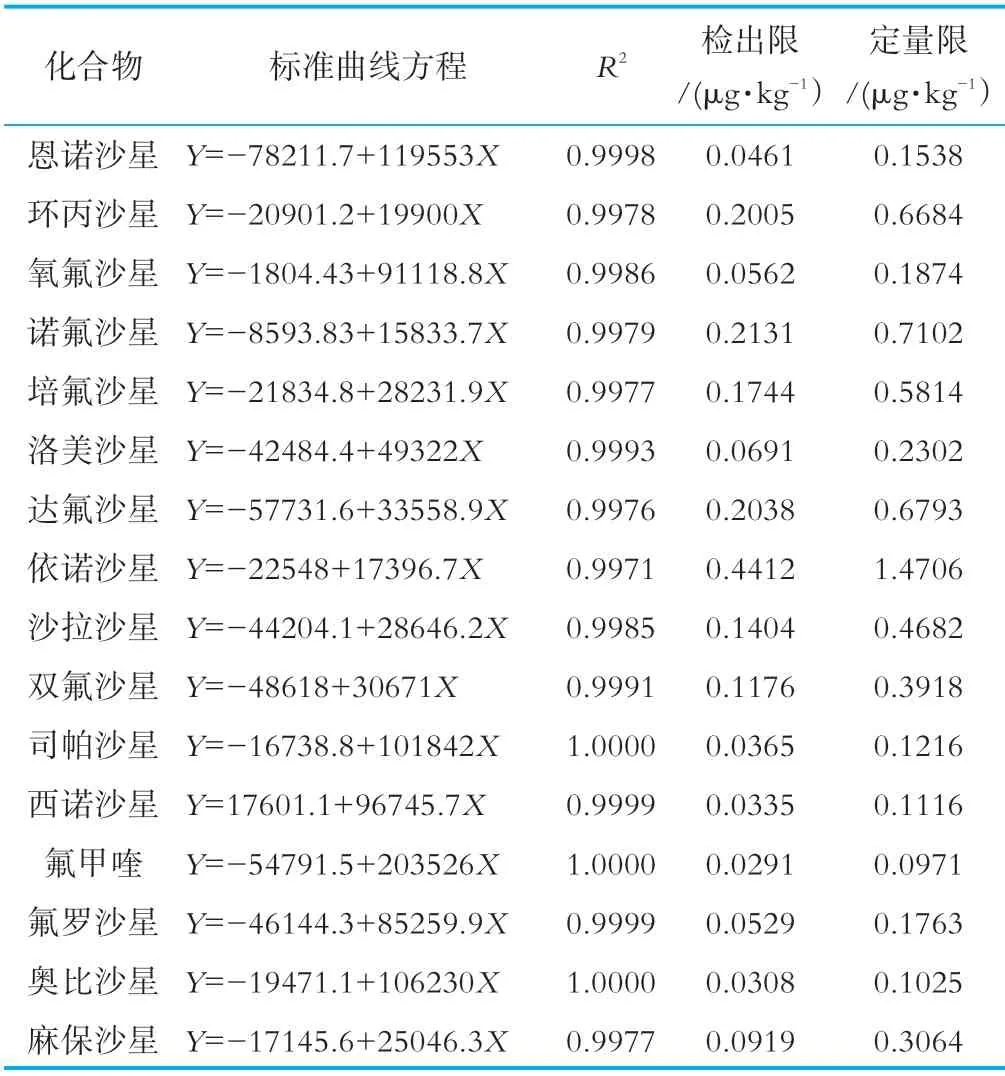
表3 喹诺酮化合物的标准曲线方程、相关系数、检出限和定量限
2.5 准确度和精密度结果分析
由表4可知,当喹诺酮加标浓度为2.0μg/kg时,回收率范围为78.7%~103.9%,相对标准偏差RSD为1.4%~3.9%;当喹诺酮加标浓度为4.0μg/kg时,回收率范围为78.1%~105.3%,相对标准偏差RSD为0.9%~8.8%;当喹诺酮加标浓度为20.0μg/kg时,回收率为78.4%~100.2%,相对标准偏差RSD为1.4%~8.4%。3个浓度条件下加标回收率和精密度良好,回收率符合GB/T 27404-2008中当加标浓度<0.1 mg/kg时,回收率范围为60%~120%。结果表明本研究所建立的方法满足奶粉中16种喹诺酮化合物的测定。

表4 喹诺酮化合物的回收率和精密度
2.6 基质效应评价
由图4可知,除依诺沙星和西诺沙星表现为基质增强效应,其他14种喹诺酮的基质效应在83.6%~119%范围内,基质效应不明显。因液相色谱-串联质谱仪的离子源结构特点,均出现基质效应,通常会对目标化合物的灵敏度、准确度和重现性等产生影响。基质效应是一种评价检验方法的有效手段。通常采用改变净化手段、增加同位素内标及基质标准曲线来降低基质效应,内标价格昂贵,且测定多种化合物需要添加多种内标,会大大提高检验成本。本研究采用分散固相萃取法净化了奶粉中的蛋白质脂肪金属元素等,并通过基质标曲降低基质效应。结果表明此研究方法中大部分喹诺酮类化合物基质效应不明显,保证检验结果准确性。
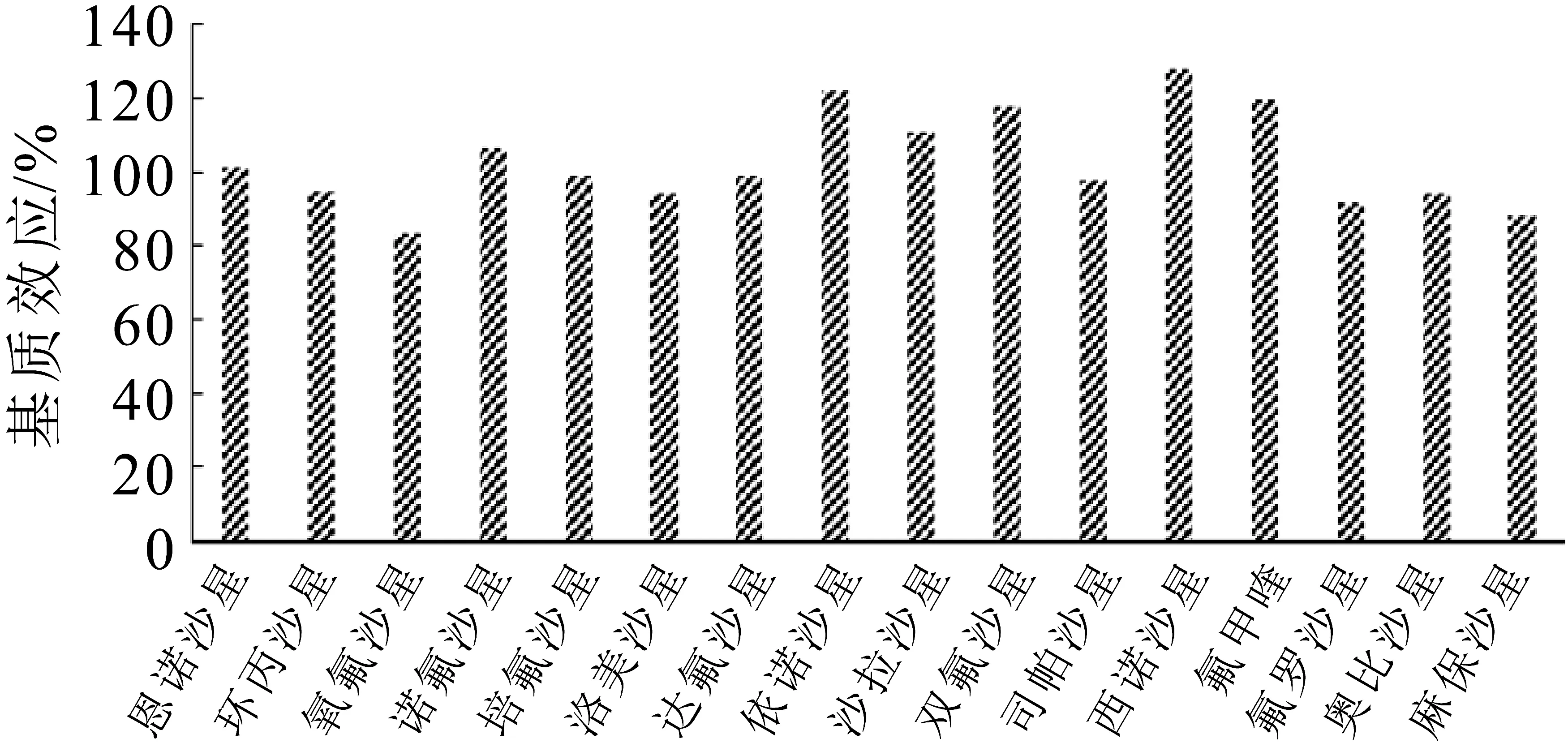
图4 16种喹诺酮化合物的基质效应
2.7 方法运用
从市场购买儿童奶粉、成人奶粉和老年奶粉、运用所建立的快速检测方法进行检测,样品中均未检出上述16种喹诺酮类化合物,且质控样品加标回收率符合GB/T 27404-2008要求,表明结果准确可靠。说明乳粉中喹诺酮化合物残留风险较小,下一步将增加样品量进行检测。
3 结论
本文建立了快速检测乳粉中16种喹诺酮类药物残留的超高效液相色谱串联质谱法。采用液液萃取法提取样品,分散固相萃取法净化样品,并对检出限、定量限、准确度等方法学指标进行了验证,采用基质效应评价了检验方法的可靠性。与相关国家标准相比,此研究验证了乳粉中喹诺酮化合物的检测方法,增加了所检测喹诺酮化合物的数量,定量限优于国家标准,采用分散固相萃取法净化样品,提高了检验效率,降低了成本,提高了检测通量,且基质效应不明显。该方法作为批量快速检测乳粉中16种喹诺酮类化合物定量检测方法,具有简单高效、灵敏度高等优点。为有效监控乳粉中喹诺酮药物残留的提供了技术支持,保证了乳制品安全。
