伴有生长迟缓的47,XXX综合征3例报道
2021-12-31冯亚琴谢理玲王荻兰
杨 利,冯亚琴,杨 玉,谢理玲,王荻兰,黄 慧
南昌大学附属儿童医院/江西省儿童医院内分泌遗传代谢科,南昌 330006
47,XXX综合征又称为超雌综合征,最早于1959年由Jacobs等[1]报道,是一种性染色体非整倍体疾病。与正常女性核型46,XX相比,47,XXX综合征患者多了1条X性染色体。丹麦细胞遗传登记中心统计的数据[2]显示,该病发生率在女性新生儿中约为11/10 000,只有接近13%的患者被确诊。47,XXX综合征的临床特征多不典型,最常见的特征包括身材高大、皮下皱褶、低眼压和斜指、癫痫发作、肾脏和泌尿生殖系统异常等,卵巢早衰也可能是相关的表现。目前,已报道的47,XXX综合征病例多表现为身材高大,身材矮小的患儿仅有1例报道[3],且国内外均无使用重组人生长激素(recombinant human growth hormone,rhGH)治疗的报道。本文回顾性分析了3例伴有生长迟缓表现的47,XXX综合征患儿的临床特征,以期为该病的临床诊断提供参考。
1 临床资料
1.1 病例1
患儿,女,10岁4个月,因发现生长速度缓慢数年伴乳房增大3个月于2016年5月29日就诊。患儿生长速度缓慢数年,年生长速率<4 cm/年;入院前3 个月出现乳房增大,无触痛;无慢性头痛、恶心、呕吐,无视物模糊,无嗅觉改变;运动和智力发育无明显落后。患儿为G2P2(第2 胎,第2 产),足月剖宫产,无产时缺氧窒息史,出生体质量2 800 g。父亲身高169 cm,母亲身高159 cm,父母均体健。入院查体:身高128.5 cm[<-2 标准差(standard deviation,SD)],体质量30 kg(P25~P50),体质量指数 (body mass index, BMI) 18.17 kg/m2(P50~P85);身材矮小,体型匀称,无特殊面容;心脏、肺、腹部查体无特殊;Tanner 分期为双侧乳房B2 期、阴毛PH1 期、腋毛A1 期。辅助检查:黄体生成素(luteinizing hormone, LH) 1.56 U/L; 卵泡刺激素(follicle stimulating hormone,FSH) 2.56 U/L;雌二醇(estradiol, E2) 39 pg/mL; 促性腺激素释放激素(gonadotrophin releasing hormone,GnRH)激发试验(戈那瑞林)示LH 峰值14.65 U/L,FSH 峰值15.41 U/L,LH/FSH比值为0.95,提示性腺轴已启动;生长激素激发试验(精氨酸、可乐定)示生长激素(growth hormone,GH)峰值8.94 μg/L,胰岛素样生长因子-1 (insulin-like growth factor-1,IGF-1)水平正常,其余均为阴性。子宫和卵巢B 超:子宫大小在正常范围内;左侧卵巢大小约为26 mm×15 mm×12 mm(根据卵巢容积计算公式[4],左侧卵巢容积2.040 mL),未见明显增大的卵泡回声;右侧卵巢显示不清。左手X 线正位片评估骨龄8 岁(Greulich-Pyle 图谱法)。肝、脾、胰、双肾、肾上腺B超未见明显异常。垂体磁共振成像未见明显异常。外周血淋巴细胞G 显带法染色体核型分析示47,XXX(图1)。建议家属完善患儿荧光原位杂交技术(fluorescencein situhybridization,FISH) 检测,家属拒绝,且之后失访。

图1 3例患儿染色体核型Fig 1 Chromosomal karyotypes of 3 children
1.2 病例2
患儿,女,7 岁7 个月,因发现生长速度缓慢3 年,于2016 年8 月24 日就诊。患儿于3 年前发现生长速度缓慢,年生长速率不详;无慢性头痛、恶心、呕吐,无视物模糊,无嗅觉改变;运动和智力发育较同龄儿无明显落后。患儿为G2P2,足月剖宫产,无产时缺氧窒息史,出生体质量3 050 g。父亲身高155 cm,母亲身高150 cm,父母均体健。入院查体:身高118.5 cm(<-2 SD),体质量30.5 kg(P50~P75),BMI 18.52 kg/m2(P85~P90);身材矮小,体型匀称,无特殊面容;心脏、肺、腹部查体无特殊;Tanner分期为双侧乳房B1期、阴毛PH1期、腋毛A1期。辅助检查:LH 1.56 U/L;FSH 0.82 U/L;E2<11.80 pg/mL;生长激素激发试验示GH 峰值14 μg/L,IGF-1 水平正常,其余均为阴性。子宫和卵巢B 超:子宫大小在正常范围内;左侧卵巢大小约为15 mm×14 mm×13 mm(容积1.429 mL);右侧卵巢大小约为16 mm×15 mm×12 mm(容积1.507 mL);双侧卵巢可见2 枚直径>4 mm的卵泡回声;骨龄5岁;肝、脾、胰、双肾、肾上腺B超以及垂体磁共振成像未见明显异常。染色体核型分析示47,XXX(图1),未予干预及治疗。患儿11岁3个月时出现月经初潮(经期规律,量适中,无痛经)。患儿现12岁7个月,身高156 cm(P50~P75)。
1.3 病例3
患儿,女,8 岁,因发现生长速度缓慢2 年,于2020年9 月4 日就诊。患儿于2 年前发现生长速度缓慢,年生长速率<4 cm/年;无慢性头痛、恶心、呕吐,无视物模糊,无嗅觉改变,无听力异常;第一个单词的发音和开始时间为17 个月,独立行走年龄为24 个月;语言表达能力稍差,理解能力欠佳;精神好,食欲欠佳,大小便如常。患儿为G2P2,足月顺产,无产时缺氧窒息史,出生体质量2 900 g;运动和智力发育较同龄儿落后。父亲身高168 cm,母亲身高152 cm,父母、姐姐均体健,母亲、姐姐染色体检查正常。入院查体:身高113.3 cm(<-2 SD),体质量18 kg(P50~P85),BMI 14.1 kg/m2(P15~P30);身材矮小,体型匀称,面容无特殊;心脏、肺、腹部查体无特殊;无脊柱侧弯,双侧肢体对称,有肘外翻,双下肢无异常,无通贯掌;Tanner 分期为双侧乳房B1 期、阴毛PH1 期、腋毛A1 期。辅助检查:LH 0.23 U/L;FSH 0.96 U/L;E2<11.80 pg/mL;抗米勒管激素(anti-Müllerian hormone,AMH) 2.3 ng/mL;生长激素激发试验GH 峰值10.7 μg/L,IGF-1水平正常,其余均为阴性。子宫和卵巢B 超:子宫大小在正常范围内;左侧卵巢大小约为12 mm×9 mm×12 mm(容积0.678 mL);右侧卵巢大小约为12 mm×11 mm×11 mm(容积1.452 mL);双侧卵巢未见直径>4 mm的卵泡回声。肝、脾、胰、双肾、肾上腺B超未见明显异常。骨龄6 岁6 个月。垂体磁共振成像未见明显异常。染色体核型分析示47,XXX(图1)。FISH 检测示XXX 染色体比例为91%,XXXX 染色体比例为1%,XX 染色体比例为8%。予rhGH 治疗,初始治疗时间是8岁1 个月,治疗剂量0.150~0.165(IU·kg-1·d-1),持续治疗8 月余,目前仍在治疗中。患儿身高增长5.7 cm,年生长速率8.55 cm/年。患儿治疗期间随访身高曲线见图2。
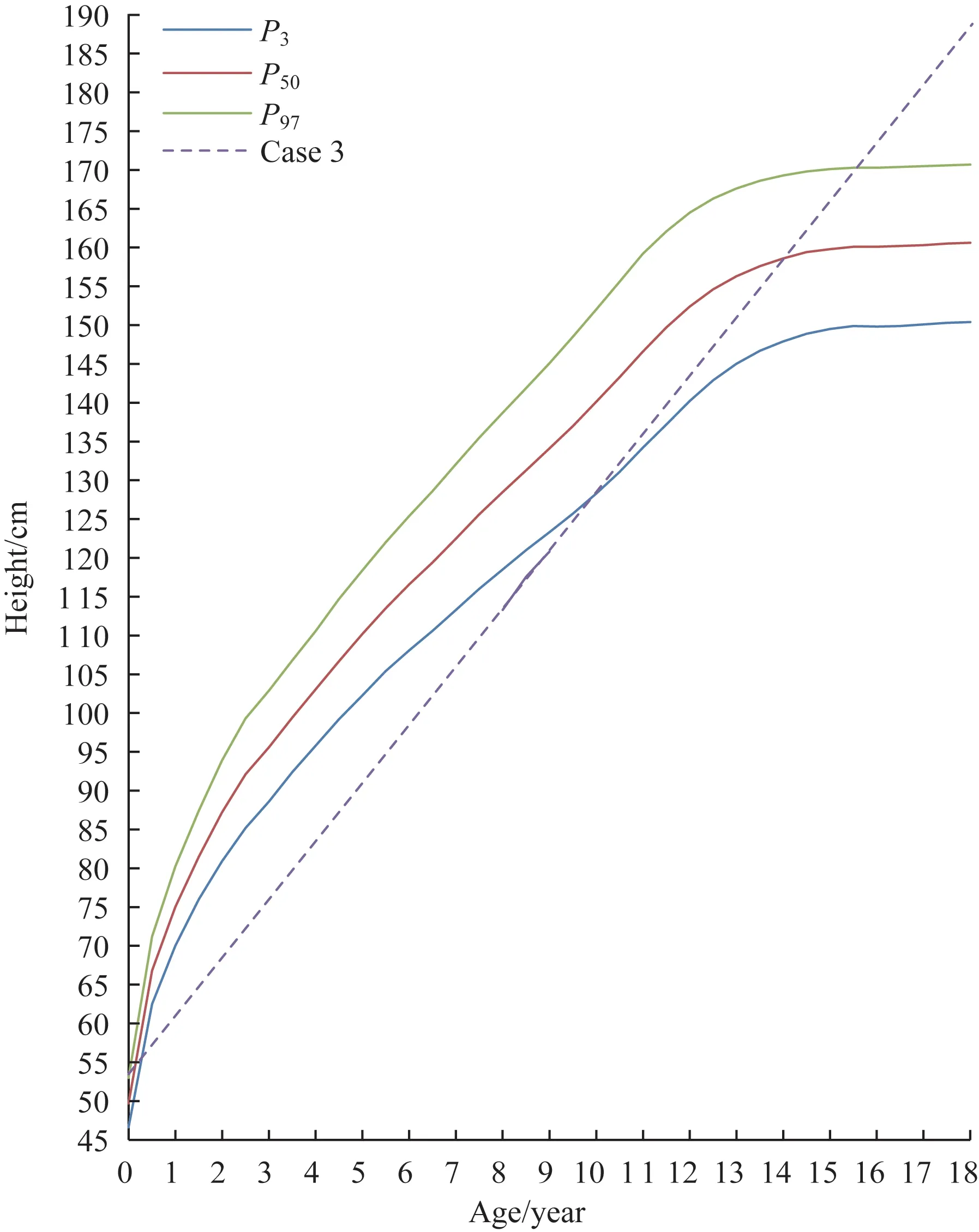
图2 病例3患儿治疗期间身高曲线Fig 2 Height curve of case 3 during treatment
2 讨论
超雌综合征是女性中最常见的性染色体非整倍体疾病,表型多样且症状较轻,缺乏明显的生理、心理异常特点。47,XXX综合征虽然有较高的发病率,但确诊率只有13%左右,诊断时的平均年龄为18.2岁[6]。在不同年龄段,超雌综合征的临床表现存在差异。青春期前(12岁前)的主要临床表现为不典型的面部畸形(唇薄、眼距过长)、低BMI、低智商、认知障碍、情绪障碍和脊柱侧凸;青春期(12~16岁)和青春期后(16岁以上)患者多表现出性腺发育不良的特征性体征(月经紊乱或生殖障碍)。超雌综合征最常见的核型是非嵌合体47,XXX;但10%的病例中可见嵌合体核型的出现,如46,XX/47,XXX等。有研究[7]发现,染色体核型中包含X染色体条数越多,精神发育迟滞、智力受损、不孕与性别分化障碍、畸形等表现越明显。超雌综合征也可合并其他疾病,包括癫痫、肾脏和泌尿生殖系统异常、先天性心脏病、小脑发育不良和枕脑膨出、系统性红斑狼疮、干燥综合征、原发性不育等[8-9]。
在典型的46,XX 女性患者中,每个细胞中只有一条X 染色体是遗传活跃的,另一条染色体是通过DNA 甲基化和整个染色体中组蛋白变体的积累而失活的。研究[10]发现X 染色体富含与智力障碍和情感障碍相关的基因。47,XXX 患者3 条X 染色体中,有2 条的特定片段即假常染色体区域(PAR1 和PAR2),具有Y 染色体同源物,不会被灭活,并保持遗传活性,且5%~10%的X 染色体上PAR区域以外的额外基因也逃脱了X失活。47,XXX患儿智商较低和精神疾病等表型可能与X 染色体过多有关。Nielsen 等[11]研究发现,47,XXX 综合征患者的X 染色体表达谱总体上与正常女性相似,但外显子上HTR2C、ZNF673等基因的甲基化减少。HTR2C在食欲和饮食行为的调节中发挥作用,可能与47,XXX 综合征患者BMI 低有关;ZNF673在神经内分泌系统的发育和分化中起主要作用,可能与低智商、认知障碍有关。
本研究分析了3例伴有生长迟缓的47,XXX女童临床表现。3例患儿均因发现生长迟缓就诊,无明显特殊面容,就诊时身高均位于-2 SD以下,出生体质量及身长均在正常范围。通过生长激素激发试验,病例1诊断为生长激素部分缺乏症,病例2、病例3诊断为特发性矮小。目前,有关超雌综合征患儿有身材矮小表现的仅见1例报道[3]。47,XXX核型的女性出生时的身高和体质量相对于同胎龄新生儿通常是正常的。到了青春期,大多数47,XXX的女性身高达到或超过正常同年龄者身高的P75,其最终身高介于正常人群的-1 SD~3 SD。笔者查阅相关文献,分析了超雌综合征患儿出现生长发育迟缓的可能原因。病例1患儿生长激素激发试验示生长激素部分缺乏,IGF-1水平正常,需考虑生长激素激发试验存在假阳性可能,并未复测。但Aksglaede等[12]研究发现47,XXX综合征患儿的IGF-1水平不能反映其生长情况。病例2、病例3患儿生长激素激发试验示生长激素峰值>10 μg/L,患儿出现生长发育迟缓的原因可能与SHOX基因单倍体不足、雌激素水平过高、骨矿物质状态和骨代谢受损、染色体核型为包含X单体和X三体的嵌合体核型的特纳综合征(Turnet syndrome,TS)有关。SHOX基因位于性染色体假常染色体区域1的远端,在骨骼发育和生长过程中起重要作用。大量研究表明SHOX基因单倍功能不足、异常突变可能导致身材矮小[13]。E2对男性和女性的骨骺成熟、正常骨骼比例和骨矿化都起着重要作用,对骨骺生长有双向作用,高浓度时使骨骺板闭合,停止线形生长。本研究中的3例患儿E2水平均在正常范围内,暂不考虑患儿早期生长发育迟缓是雌激素水平过高所致。Stagi等[14]研究发现,超雌综合征患者可能存在骨矿物质状态和骨代谢受损;Frederiksen等[15]同样发现SHOX基因的突变可能对骨骼微结构产生影响。因此,并不能排除骨代谢异常是3例患儿生长发育迟缓的原因。Butnariu等[16]对36例超雌综合征患儿资料进行回顾性研究,对外周血淋巴细胞G显带法染色体核型分析结果为47,XXX的患儿,完善其口腔黏膜涂片X染色质(Barr小体)分析,发现55.55%的患儿为包含X单体和X三体的嵌合体核型的TS,如45,X/47,XXX等;并且发现包含TS 细胞系的嵌合体表型与细胞系的类型相关,表现多以TS 体征为主,即出现身材矮小、特殊面容等,精神障碍、智力低下等表现少见,且较普通TS 体征轻微,如心血管异常、肾脏异常、先天畸形(<5%)以及骨骼异常(<4%)的发生率偏低。Wallerstein等[17]对身材矮小和TS表型的女性进行核型检测,同样发现47,XXX细胞系存在于5%~15%的TS患者中。因此,当发现患有47,XXX并伴有TS 表型的个体,应完善细胞遗传学、口腔黏膜涂片X染色质(Barr小体)检测或FISH检测。本研究中,病例3患儿完善FISH检测后仍考虑为47,XXX综合征;但病例1、病例2患儿未完善FISH检测,故不能排除其生长发育迟缓原因为染色体核型包含TS 细胞系在内的嵌合体。
病例3予rhGH治疗后,年生长速率达8.55 cm/年,生长速度较前增加>3 cm/年;与生长激素缺乏症患儿使用rhGH后年生长速率(10~12 cm)/年相比,治疗效果较好。目前,国内外并无超雌综合征患者使用rhGH的相关报道。病例3患儿仍在治疗中,笔者会继续观察患儿后续情况。病例3患儿AMH值偏低。Davis等[18]对15例非嵌合体超雌综合征患儿进行AMH分析,发现2/3的超雌综合征女童AMH水平低于正常值。AMH可反映卵巢储备功能,47,XXX女童和青少年卵巢储备减少,可能表明这些患者最终发生卵巢早衰(premature ovarian insufficiency,POI)的风险增加。POI的潜在病因,包括导致卵巢发育异常或局限性卵巢自身免疫的先天性畸形,X染色体上POI候选基因(POF1B、STS、XNPEP2、FMR-1、USP9X)的调控中断或突变、重复或缺失,X染色体上促凋亡基因(AIFM1)的过度表达或抗凋亡基因(BCORL1)的表达不足等。
目前,对于超雌综合征尚无特效治疗方法,应多学科合作、共同管理,以对症治疗为主。对患儿应详细询问其病史,早期进行全面的发育评估,包括语言、运动和个人-社交能力方面。当评估分数低于参考值时,应考虑完善染色体核型分析,早期干预,定期至儿童保健科、儿童心理行为科、内分泌科、神经内科随访,进行智力发育评估,并给予相应治疗。对婴幼儿,应定期复查肾脏彩色超声、心脏超声、脑电图等。青少年女性出现月经初潮延迟、月经不调或生育问题时,应完善性发育水平相关检查,进行卵巢早衰评估。研究[19]发现超雌综合征的预后取决于表现的严重程度以及治疗的质量和时机;与出生后诊断的女性患儿相比,产前诊断的女性患儿精神障碍发生率更低。
综上所述,对于不伴有特殊面容的生长发育迟缓女童,在完善生长轴相关检查的同时,应完善染色体核型分析及检测,排除染色体疾病,考虑TS 的同时应排除有无超雌综合征可能,避免漏诊与误诊,为进一步治疗提供支持。
