LC-MS/MS法测定人血浆中利奈唑胺浓度的不确定度评定
2021-12-30刘艳芳张琛琛蔡朝红党大胜北部战区总医院临床试验研究室沈阳0840沈阳药科大学生命科学与生物制药学院沈阳0840北部战区总医院临床药学室沈阳0840
刘艳芳,张琛琛,蔡朝红*,党大胜(.北部战区总医院临床试验研究室,沈阳 0840;.沈阳药科大学生命科学与生物制药学院,沈阳 0840;3.北部战区总医院临床药学室,沈阳 0840)
利奈唑胺(linezolid)是第一个应用于临床的唑烷酮类全合成抗菌药,临床主要用于治疗耐万古霉素屎肠球菌引起的感染、耐甲氧西林金黄葡萄球菌引起的院内获得性肺炎等[1]。由于利奈唑胺独特的作用机制与现有抗菌药无交叉耐药,被认为是治疗多重耐药革兰氏阳性菌感染的最后防线[2]。随着利奈唑胺应用的增多,一些较为严重的不良反应逐渐暴露出来[3-4],临床对其在中国人体药动学研究以及特殊人群实时的血药浓度监测的需求越来越高,基于此本研究室建立了高效液相色谱-串联质谱(LC-MS/MS)法测定人血浆中利奈唑胺浓度,同时为了控制检测质量,达到检测数据国际互认的要求对方法进行不确定度评价。
1 材料
UFLC-20AD超高速液相色谱系统(日本岛津公司),3200Q-Trap质谱仪(美国AB Sciex公司),Mettler MS 105分析天平(Mettler),Eppendorf 5804R低温冷冻离心机(Eppendorf),IKA VORTEX GENIUS 3 mm-1型振荡器(IKA VORTEX)。利奈唑胺(批号:130640-201901,纯度:99.6%,规格:100 mg/支),卡马西平(批号:100142-201706,纯度:100.0%,规格:100 mg/支)(中国食品药品检定研究院),甲醇(批号:1099307025,色谱纯,德国MERCK公司),超纯水由Milli Q 型纯水仪制得(Millipore,USA)。
2 方法
2.1 分析条件[5-6]
色谱柱:Waters Xterra MS C18(3.5 μm,100 mm×2.1 mm),流动相:A为纯水,B为甲醇,梯度洗脱:0~2.0 min,5%~95%B;2.0~3.0 min,95%B;3.0~3.1 min,95%~5%B;3.1~5.0 min,5%B。流速:0.35 mL·min-1,柱温:40℃,进样量:0.5 μL。电喷雾离子源;正离子模式;多反应离子监测方式;喷射电压:4500 eV;源温度:550℃;离子源气体:50 psi;辅助气:25 psi;气帘气:35 psi;碰撞气:medium;扫描时间:150 ms;定量分析离子对:338.2~296.1(利奈唑胺),237.0~194.0(卡马西平)。
2.2 溶液配制
2.2.1 系列对照品溶液 精密称取利奈唑胺对照品22.13 mg置10 mL量瓶中,甲醇溶解并定容,得2.20 mg·mL-1储备液。精密移取上述储备液适量用甲醇逐级稀释,即得2.00、5.00、20.0、50.0、100、200、400 μg·mL-1的系列对照品溶液以及质量浓度为4.00、40.0及320 μg·mL-1的低(QCL)、中(QCM)、高(QCH)质控溶液。
2.2.2 内标溶液配制 精密称取卡马西平10.79 mg置10 mL量瓶中,甲醇溶解并定容,得1.08 mg·mL-1内标储备液。精密移取23.2 mL上述内标储备液置10 mL量瓶,甲醇定容得质量浓度为2.50 μg·mL-1的内标溶液。
2.3 校正标样/质控样品的制备
取对照品溶液/质控溶液10 mL置1.5 mL离心管中,分别加入90 mL人空白血浆、10 mL内标溶液、700 mL甲醇,涡旋混合1 min,4℃离心10 min(1610×g),取上清液50 mL,加甲醇150 mL涡旋混合1 min,即得。
2.4 数学模型的建立
yi=axi+b,其中yi为待测物响应信号与内标响应信号之比,a为标准曲线斜率;b为标准曲线截距;xi为待测物浓度与内标浓度之比。
3 结果
3.1 不确定度来源
样品测定的每一个步骤都有可能引入不确定度,本实验测量不确定度来源主要包括操作重复性、称量、溶液配制、提取回收、基质效应、仪器量化、标准曲线拟合等。
3.2 不确定度评定
3.2.1 重复性引入的不确定度Ur(1) 取质控样品QCL、QCM、QCH各6份,按“2.3”项下方法处理后,进样分析,计算质控样品浓度,重复进行3组,结果见表1。
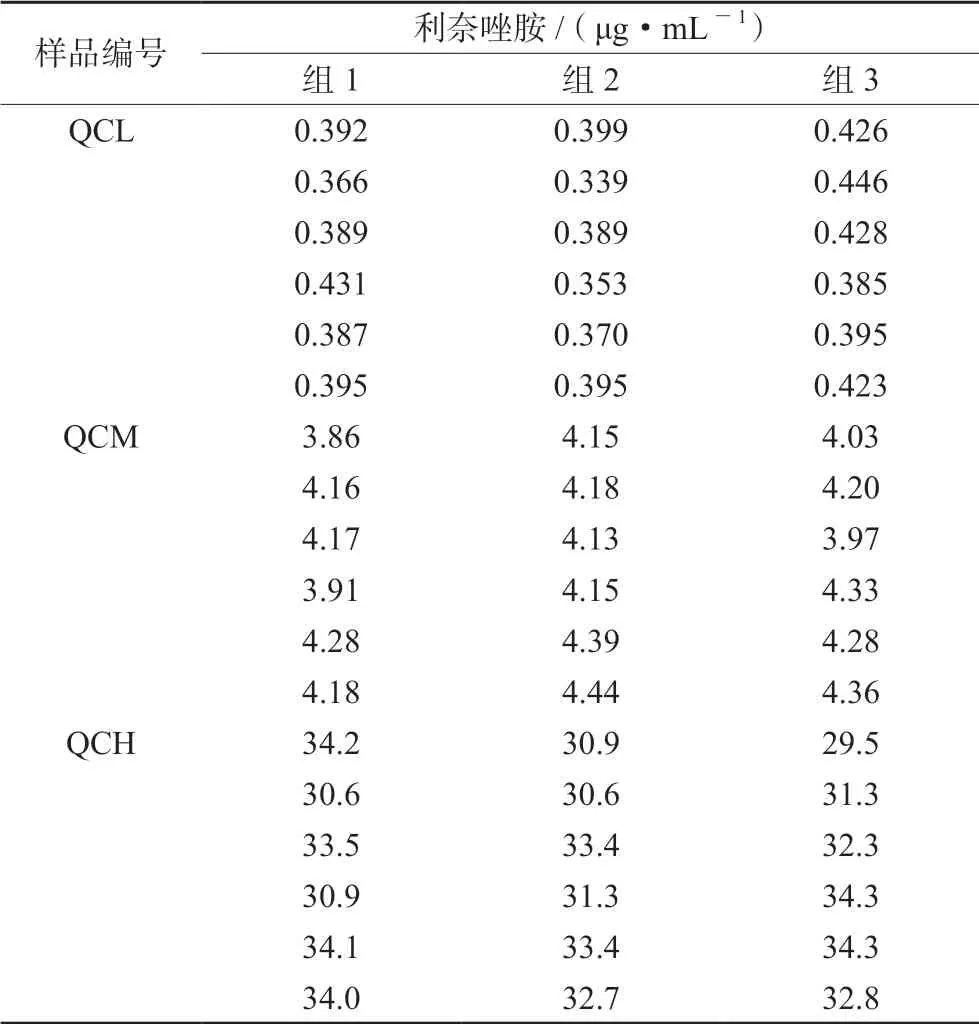
表1 利奈唑胺重复测定数据 Tab 1 Repeated measurement of linezolid concentration
依据贝赛尔公式计算利奈唑胺合并样本偏差:
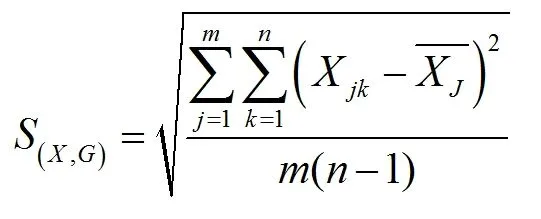
公式中k为每组平行测定次数(1,2,...,n),j为组数(1,2,…,m),G为组别(L、M、H)。
经计算:S(X,L)=0.023 μg·mL-1,S(X,M)=0.156 μg·mL-1,S(X,H)=1.614 μg·mL-1
以每组平均值表示测量结果,平均值的标准偏差公式为:
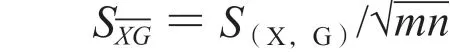
经计算:
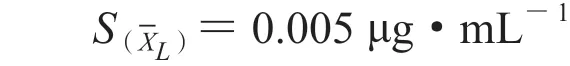
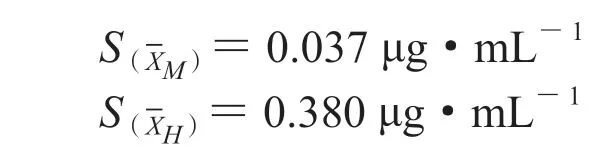
测量结果的相对标准不确定度为:

经计算:Ur(1,L)=0.0136,Ur(1,M)=0.0088,Ur(1,H)=0.0117。
3.2.2 称量引入的不确定度,Ur(2) 称量过程,天平灵敏度带来的不确定度可忽略不计,依据计量检定证书,所用电子天平e=0.01 mg。按矩形分布考虑,包含因子k=,随机变量半宽a=0.5e,天平的非线性误差为:
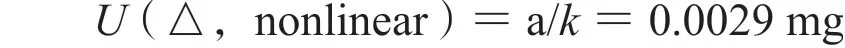
天平自动调零引起的误差,其 a0=a,则天平自动调零引起的误差为:
不考虑重复性误差时,天平的标准不确定度为:
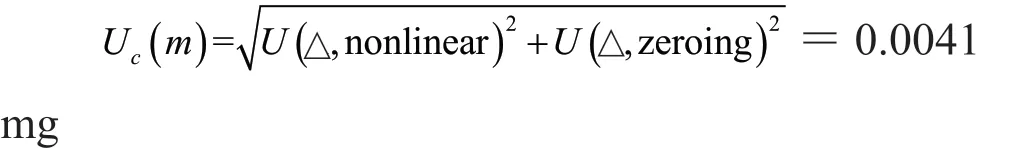
利奈唑胺的称量相对标准不确定度为:
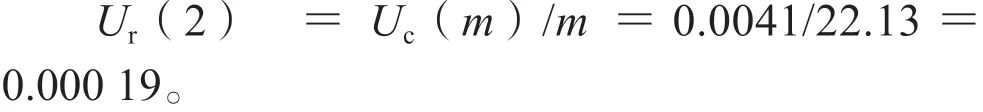
3.2.3 溶液配制引入的不确定度,Ur(3)
(1)对照品纯度引入的不确定度,此项属B类不确定度,按矩形分布处理,其相对标准不确定度如下:
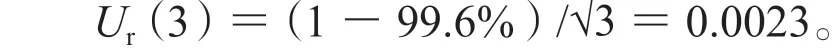
(2)标准溶液及质控溶液配制过程引入的不确定度:配制溶液用到的移液枪为Thermo Finnpipette F3移液枪,型号为 2~20 μL(P1)、10~100 μL(P2)、20~200 μL(P3)和 100~1000 μL(P4),其容量的最大允差分别为±0.2 μL、±0.8 μL、±1.6 μL和±8 μL,10 mL量瓶(V1)最大允差为±0.020 mL。Ur(P1)=a/() =0.0058,Ur(P2)=Ur(P3)=Ur(P4)=0.0046,Ur(V)=0.000 57。
利奈唑胺系列标准溶液配制过程共使用10 mL量瓶(V1)1次,移液枪2~20 μL(P1)3次,10~100 μL(P2)1次,20~200 μL(P3)1次,
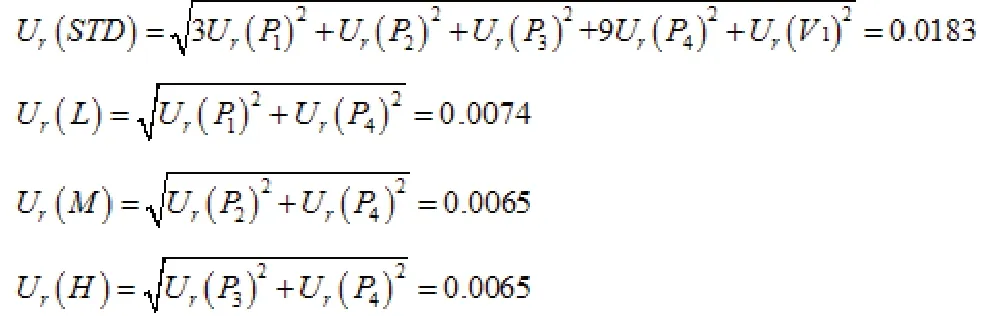
则利奈唑胺溶液配制整个过程引入的相对标准不确定度为:
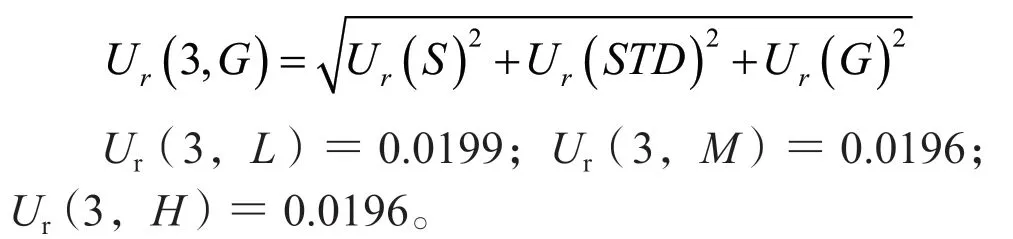
3.2.4 校正标样和质控样品制备引入的不确定度,Ur(4) 校正标样(S)和质控样品(G)均按照“2.3”项下方法制备,使用移液器及次数为:2~20 μL移液器2次,10~100 μL移液器2次,20~200 μL移液器1次,100~1000 μL移液器1次。标准曲线共6个点,则样品相对标准不确定度为:
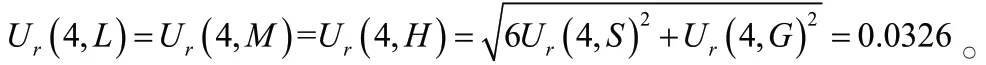
3.2.5 样品提取过程引入的不确定度,Ur(5) 按“2.2.1”项下方法制备低、中、高浓度质控样品,每浓度6份并进样分析,得到与内标的峰面积比值为C;取18份空白血浆,加入700 μL甲醇,混匀,4℃离心10 min(1610×g),取上清液并加入标准溶液和内标溶液使其浓度与质控样品浓度相同,涡旋混合1 min后取上清液50 mL,加甲醇150 mL,涡旋混合1 min后取0.5 mL进样分析。每浓度6样本分析,获得相应峰面积为B,则利奈唑胺在血浆样本中的提取回收率=C/B×100%,低、中、高浓度质控样品回收率分别为(94.4±9.36)%、(98.1±1.87)% 和(102.5±4.12)%。回收率的相对标准不确定度计算公式为:
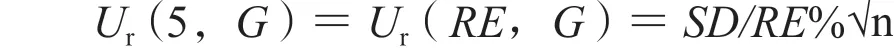
则,利奈唑胺低、中、高浓度质控样品回收率的相对标准不确定度分别为:

3.2.6 基质效应引入的不确定度,Ur(6) 取处理后基质及空白溶剂配制低、高浓度质控样品,每浓度平行测定6次,内标归一化基质效应=处理后基质配制样品的峰面积与内标峰面积比值(A)/空白溶剂配制样品的峰面积与内标峰面积比值(B),计算得低、高浓度质控样品的基质效应分别为(117.1±4.64)% 和(115.5±5.75)%,基质效应引入的相对标准测量不确定度计算公式为:则利奈唑胺低、高浓度质控样品的相对标准不确定度为:
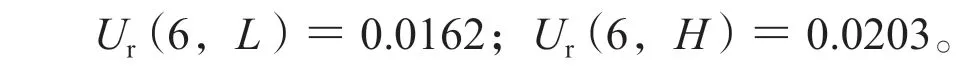
3.2.7 仪器量化引入的不确定度,Ur(7) 所用液质联用仪为AB3200 Q-Trap,其中质谱定量的最大允差为2%,液相色谱取样的最大允差为1%,按矩形分布,仪器量化的相对标准不确定度为:
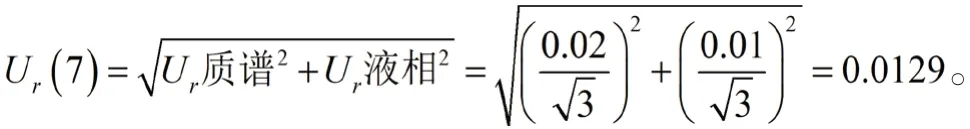
3.2.8 线性拟合引入的不确定度Ur(8) 本实验标准曲线除零点外有6个浓度点,连续测定6次,每个样品中利奈唑胺峰面积与内标峰面积的比值见表2,经拟合曲线回算校正标样中利奈唑胺浓度结果见表3。
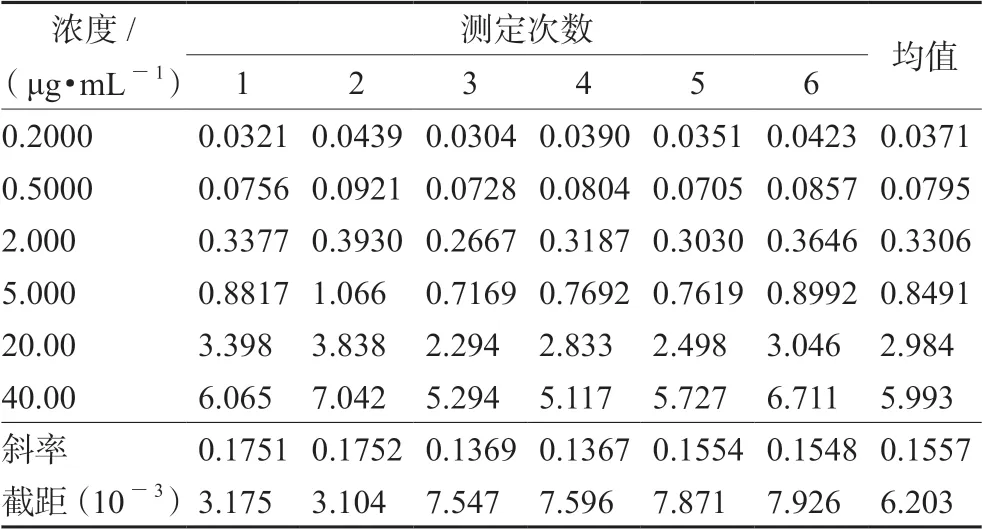
表2 利奈唑胺与内标峰面积比及回归曲线参数 Tab 2 Peak area ratio of linezolid with the internal standard and parameters of the calibration curves
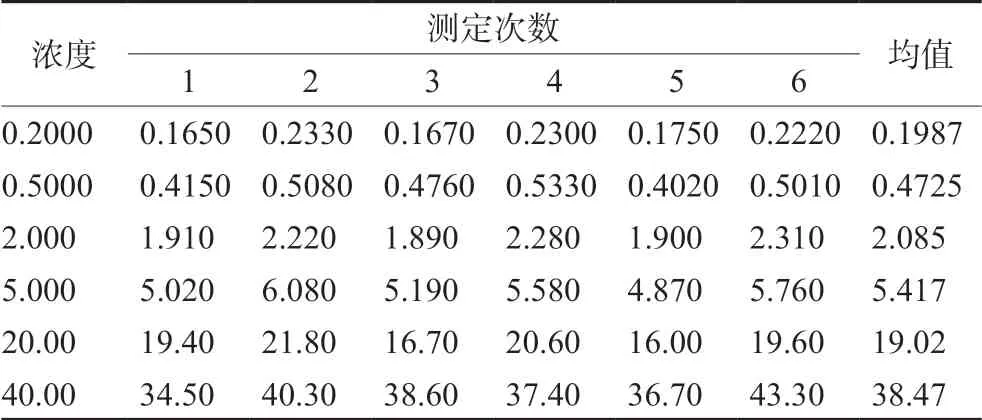
表3 经拟合曲线计算出的每个校正标样中利奈唑胺的浓度 (μg·mL-1) Tab 3 Back-calculated concentration of linezolid in standard plasma samples (μg·mL-1)
用生物样品峰面积比的平均值对浓度以加权最小二乘法进行线性回归(权重1/x2)得回归方程,斜率a=0.1557,截距b=6.203×10-3。每个浓度重复分析的次数为6次,m=6;标准曲线有6个浓度,n=6;N为测定标准血浆溶液的总次数,N=m×n=36,共测定36次;j为测定标准血浆的序数(j=1,2,3…N);为6个标准血浆浓度的理论平均值,=11 μg·mL-1。
残余标准偏差为:
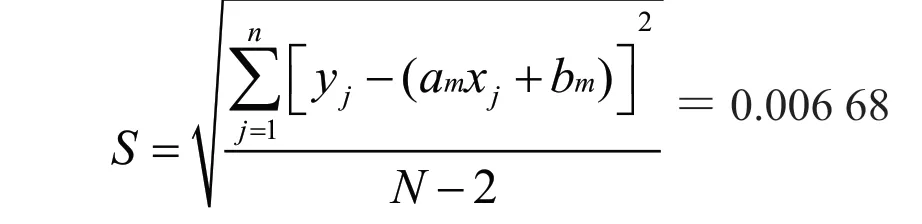
用本方法测定低、中、高浓度质控样品各18次(P=18),得到平均浓度
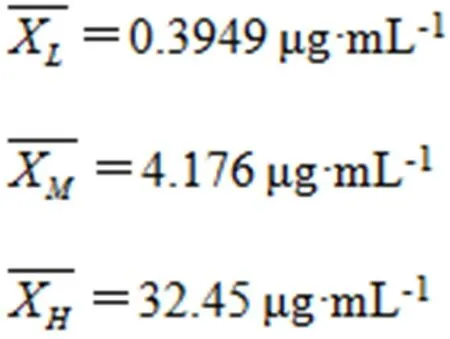
其标准不确定度为:
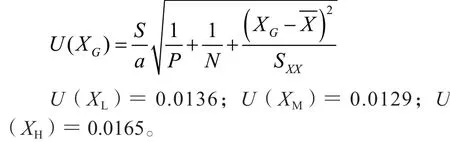
按公式计算利奈唑胺低、中、高浓度质控样品相对标准不确定度为:

则:Ur(8,L)=0.0344;Ur(8,M)=0.0031;Ur(8,H)=0.0005。
3.2.9 其他因素引入的不确定度 本实验操作中间步骤少,过程简单,耗时短,且样品制备室、进样室等均有空调进行温度控制,操作过程几乎无温度波动,故忽略温度影响。样品制备过程取液前均进行了充分震摇,有效避免取液的不均一性,因此本文忽略不均一性带来的不确定度。
3.3 合成不确定度的评定
3.3.1 标准测量不确定度的合成 依据不确定度传播律对各相对标准测量不确定度进行合成:
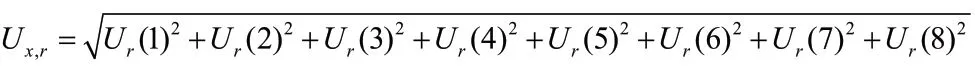
则利奈唑胺质控样品的合成相对标准测量不确定度分别为:
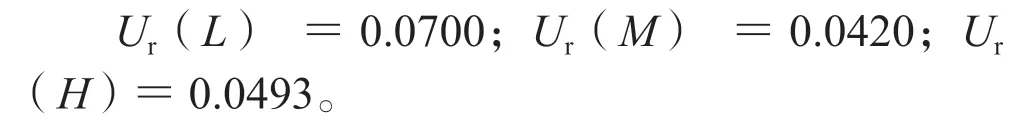
利奈唑胺质控样品测定的合成标准测量不确定度为:
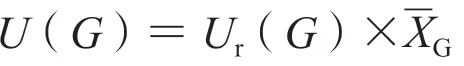
则:U(L)=0.0276;U(M)=0.1753;U(H)=1.6011。
3.3.2 标准测量不确定度的扩展 用简易评定方法,取k=2,此时对应的置信概率P=95%,得到扩展不确定度为:UG=k×U(G),则:UL=0.055;UM=0.351;UH=3.20。
3.4 测定结果的表示
人血浆中利奈唑胺低、中、高浓度质控的测定结果在k=2(P=95%)时可以分别表示 为(0.395±0.055)μg·mL-1、(4.18±0.351)μg·mL-1和(32.5±3.20)μg·mL-1。
4 讨论
本文依据JJFl059-1999《测量不确定度评定与表示》[7],中国合格评定国家认可委颁布的CNAS-CL01-G003:2019《测量不确定度的要求》[8]和CNAS-GL006:2019《化学分析中不确定度的评估指南》[9],参照已发表文章的方法[10-13],按照实验过程寻找不确定度的来源,各不确定度分量的统计直方图见图1,由图可知低浓度质控样品不确定度来源主要为提取回收、线性拟合和样品制备,中浓度不确定度来源主要为样品制备,高浓度不确定度来源主要为样品制备、基质效应和溶液配制。
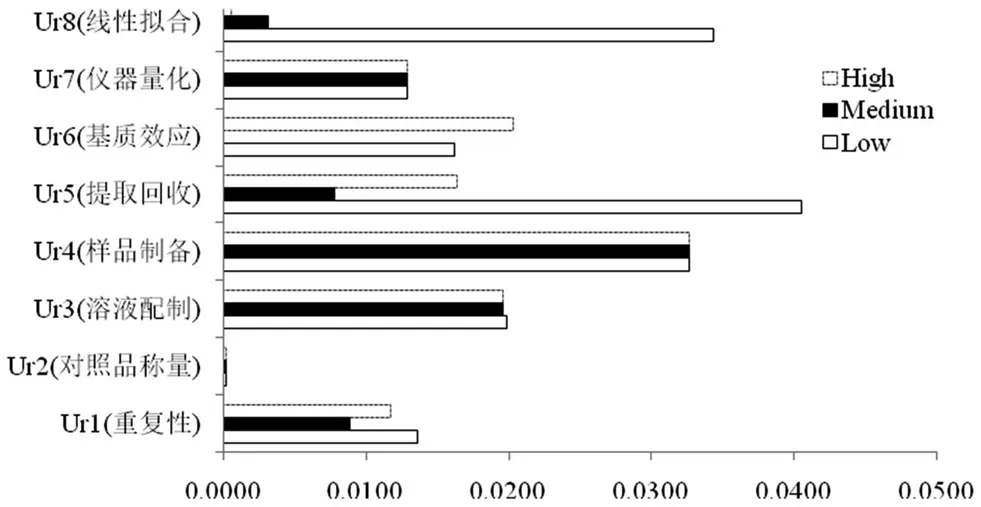
图1 液质联用法测定利奈唑胺不确定度分量统计直方图Fig 1 Histogram of uncertainty components in linezolid determination by LC-MS/MS
本文采用甲醇沉淀蛋白法进行样品提取,处理过程简单,干扰物质少,且方法无残留效应,对比中、高浓度质控样品不确定度结果,低浓度不确定度高主要因为SD值较高,即操作不平行导致,增加操作培训及考核可有效避免此类问题。线性拟合对低浓度样品的不确定度贡献较大,这与同类文献[10]结论一致,本文在不改变线性范围的情况下,对标准曲线的点数进行考察,分别用5、6、7个点作标准曲线,线性均合格的情况下,不确定度值大小顺序为:5点标曲>7点标曲>6点标曲,因此本文采用6个点浓度做标准曲线,同时证明相同线性范围情况下并非浓度点数越多越好,而要根据实际情况进行考察以减少线性拟合引入的不确定度。
样品制备对低、中、高浓度质控样品不确定度贡献均较大,此项由校正样品和质控样品两个分项共同构成,按照公式标准曲线点越少,样品制备过程越简单此项值越低,但标准曲线点个数设置会影响线性拟合值,要进行综合评估来决定,可以通过优化操作过程减少此项值。
基质对高浓度质控不确定度影响较低浓度大,主要是操作平行性的影响,同时若能采用稳定同位素做内标,必要时改用其他离子源来降低基质效应从而减少不确定值。因2020年版《中国药典》只要求对低、高浓度质控样品进行基质效应考察,故本文未将中浓度纳入基质效应的不确定度评价。溶液配制由对照品纯度、对照品溶液配制及质控溶液配制三分项共同构成,可以通过提高对照品纯度,减少溶液配制体积来控制不确定度结果。
