健康青年人肠道菌群的分布特征研究
2021-12-30田学梅王慧梁静宣王耘北京中医药大学中药信息工程研究中心北京102488
田学梅,王慧,梁静宣,王耘(北京中医药大学中药信息工程研究中心,北京 102488)
人类肠道内存在大量的微生物,在参与维持正常生理过程的同时,与疾病的发生发展也息息相关[1]。肠道微生物不仅可以调控肥胖[2],干预多糖和纤维素在体内消化吸收[3],还影响胃肠道疾病如炎症性肠病、心血管疾病如高血压、代谢类疾病如肥胖、自身免疫性疾病等[4-6]。微生物组菌群结构的变化与人类健康和疾病的发生发展密切相关,利用肠道菌群的分布差异进行个体疾病、健康等的相关研究已逐渐增多。例如2型糖尿病患者与健康个体相比,其肠道内双歧杆菌数量减少[7],胆汁淤积性肝病患者与健康对照者相比,肠道菌群存在特征性改变,韦荣氏球菌属(Veillonella)相对高丰度表达与胆汁淤积性肝病强相关[8]等。肠道菌群的状态会以各种方式反馈给宿主[9],健康个体在不同生活方式、药物、饮食、疾病状态、地域以及年龄阶段,其肠道菌群的分布存在很大差异[10-11]。通过研究不同健康人的肠道菌群的分布特征,调理肠道菌群的状态以改善健康状况。因此是否可以利用不同健康人群间肠道菌群的分布差异对健康人群进行分类,以此来评价个体健康状态并对个体健康进行有效精准的健康管理呢?基于以上思考,本研究利用16S rRNA技术[12]获得的肠道菌群种层面相对丰度数据并转换为定性数据,聚类分析将健康青年人群进行分类,根据不同分类的健康青年人群间的差异菌通过数据挖掘方法[13-14],建立以不同健康青年人分类间肠道菌群的分布差异为指标的健康青年人群分类模型,并探讨不同分类的健康青年人肠道菌群分布特征。以期为青年个性化的健康管理研究奠定基础。
1 数据获得
1.1 样本来源
选择就读于北京中医药大学大一、大二的本科生4000人,随机抽样后共纳入研究对象114人,男女性别比例1∶1,年龄18~22岁。研究经学校伦理委员会批准通过,所有研究对象均了解并签署项目知情同意书。
1.2 纳入及排除标准
纳入标准:健康大学生且正常饮食无明显饮食偏颇情况。排除标准:① 患有溃疡性结肠炎、克罗恩病等炎症性肠病及慢性便秘、慢性腹泻等其他肠道疾病患者;② 使用抗菌药物、进行阑尾手术等的患者;③ 合并急性冠脉综合征、急性脑血管意外等危急重症者;④ 有严重情绪或精神异常而无法配合者;⑤ 重大疾病患者。
1.3 肠道菌群数据的收集与整理
受试者均无严重疾病,入组后前一个月避免抗菌药物、胃肠道药物等使用并正常饮食,取受试者的早餐前粪样,通过16S rRNA 测序[15]技术进行肠道微生物菌种的组成测定,获得研究样本肠道菌群种层面相对丰度数据。将肠道菌群种层面相对丰度数据转变为定性数据(相对丰度>0即定为含有这种菌,用1表示;没有相对丰度即定为不含这种菌,用0表示)。
2 方法及原理
2.1 肠道菌群定性数据的聚类分析方法
聚类分析(clustering analysis)[13]是数据挖掘中的一个重要部分,属于无监督的学习方法。根据研究目的,本文所选用的是凝聚聚类算法中的离差平方和法[16],采用欧氏距离作为聚类分析的指标,对“1.3”项下肠道菌群种层面定性数据进行聚类分析,聚类分析工作基于软件Orange 2.7实现。
离差平方和方法:如果有两个样本类G1、G2,n1、n2分别为G1、G2样本个数

若G1,G2内部点与点距离很小,则它们能很好地各自聚为一类,并且这两类又能充分分离(D12很大),这时D就很大。
2.2 聚类分析结果差异菌的筛选
基于“2.1”项下的结果发现,114名样本的肠道菌群种层面组成个体间有显著差异。为了提高后续人群分类模型构建时建模数据的质量,加快挖掘速度并且使挖掘出的规则更准确易懂,本研究分析了受试样本在不同分类时组间的差异菌,并对其进行初步筛选得到人群分类的属性指标。
利用肠道菌群种层面相对丰度数据对不同组间进行差异菌分析。当聚类数为2,通过Welch’st-test统计分析方法进行两组差异菌的分析(P<0.05);当聚类数为3~5时,通过方差分析进行多组间差异菌的分析(P<0.05)。不同分类间差异菌的筛选通过STAMP软件进行[17]。
2.3 基于差异菌的数据建立人群分类模型方法
首先将聚类后每类样本差异菌的定性数据及属性指标分别作为模型输入指标,再分别利用朴素贝叶斯(NB)算法[18]、K最近邻(IBK)算法[19]、决策树(J48)算法[20]、随机森林(RF)算法[21]进行模型的构建,并依据曲线下面积(AUC)选出优的算法及分类,为后续模型的优化奠定基础。将各分类样本集随机划分70%作为训练集,30%作为测试集。训练集用于构建模型,同时运用Cor(Correlation-based Attribute Evaluation)[22]属性筛选算法进行筛选,优化训练模型;测试集用于验证模型,最终得到基于肠道菌群的人群分类模型,具体模型构建思路如图1所示。

图1 基于肠道菌群的人群分类模型构建方法Fig 1 Construction of population classification models based on the intestinal flora
本文中涉及分类算法和特征属性选择算法均在10折交叉的环境下运行,交叉验证是数据建模中常用且有效的一种内部验证方法,常用来测试模型或算法的准确性[23-24]。所有操作均通过数据挖掘平台软件Weka 3.8实现;运行过程中所有参数均采用平台默认的参数设置。
2.4 模型评价指标
在本文中使用ROC的AUC,为ROC曲线提供了量化值,可以通过比较AUC值的大小来评估分类模型的性能[25-27]。
3 结果与讨论
3.1 肠道菌群在门、种水平的微生物含量组成分析
在门水平上,研究对象的微生物含量组成如图2所示:最终将所有的序列鉴定为22个门,其中拟杆菌门(Bacteroidetes)、硬壁菌门(Firmicutes)、变形菌门(Proteobacteria)、放线菌门(Actinobacteria)的含量较高,占测序序列总数的97%以上,其他菌门如梭杆菌门(Fusobacteria)、疣微菌门(Verrucomicrobia)、无壁菌门(Tenericutes)、TM7、黏胶球形菌门(Lentisphaerae)等的含量较低。
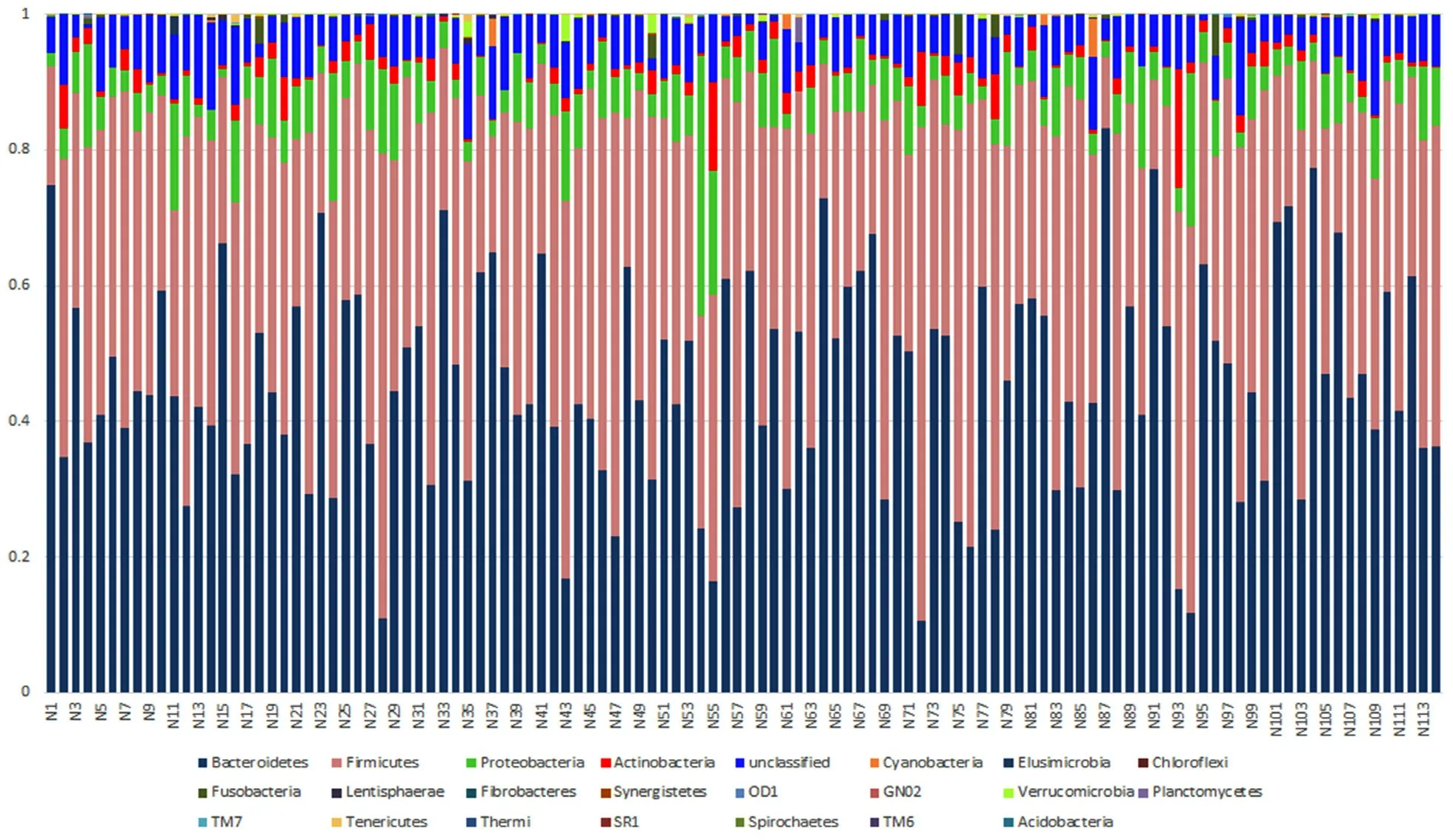
图2 门水平微生物含量组成Fig 2 Composition of microbial content at the phylum level
个体肠道菌群的组成在种水平上具有很大的差异,研究中的种水平的肠道菌群组分构成,最终将序列鉴定为257个种,相对丰度比例最高的前50个种构成的组分柱状图见如图3。
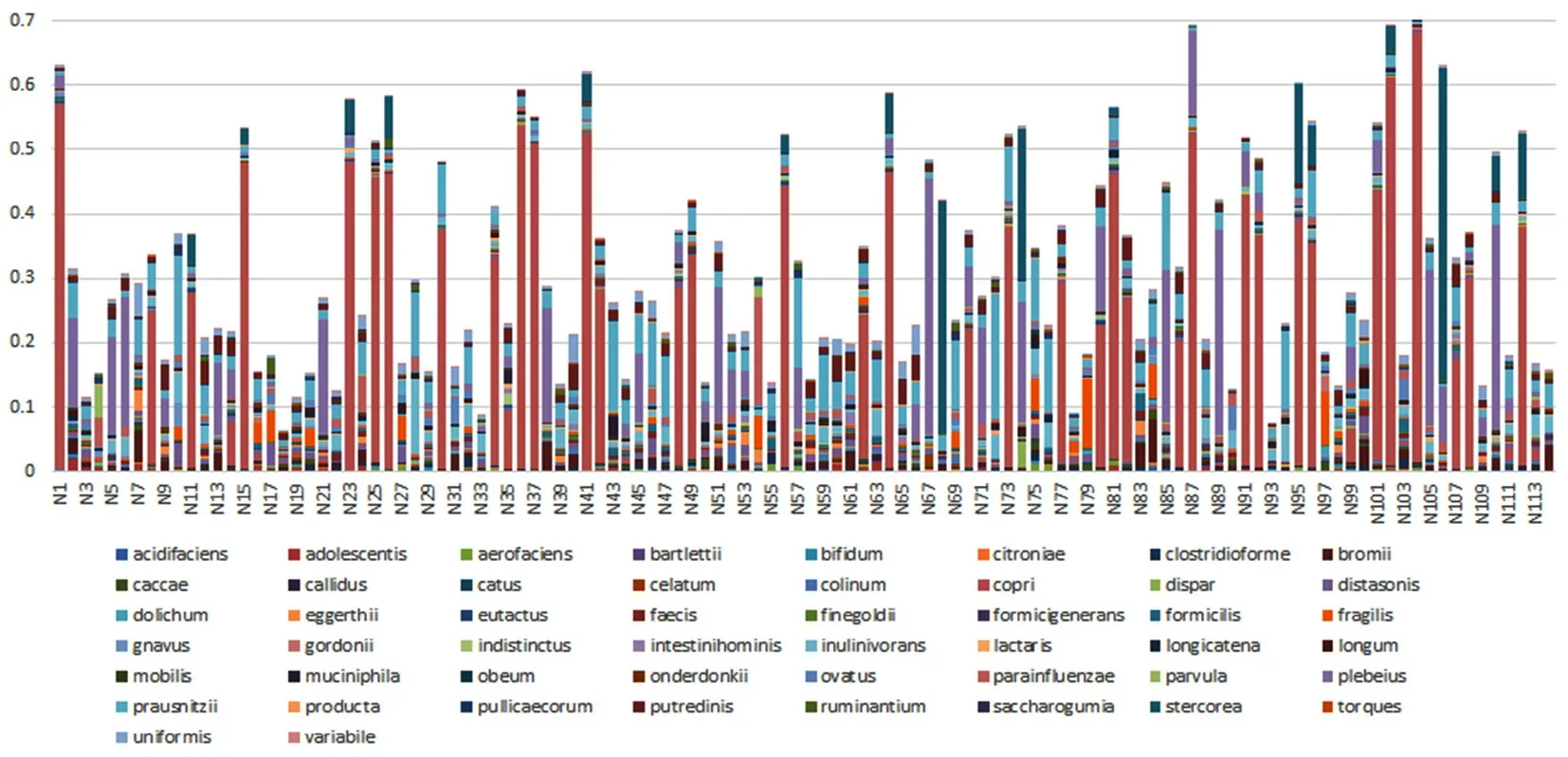
图3 受试者在种水平微生物相对丰度组成Fig 3 Relative abundance composition of subjects at the species level
3.2 肠道菌群定性数据的聚类分析
目前的聚类算法大多数需要预先给定聚类数,再对样本进行聚类分析。确定最佳聚类数是聚类分析中的关键步骤。因此为了确定最佳的聚类数,保证数据集的划分和评判结果是有效正确的,本研究依据肠道菌群种层面定性数据将受试人群划分为2~5类的结果,通过后续建立人群分类模型的结果选择最终的聚类数。聚类结果见表1。

表1 样本分为两至五类时聚类结果 Tab 1 Clustering of categories when divided into two to five categories
3.3 聚类分析结果差异菌的筛选
针对“3.2”项下不同分类人群差异菌的分析,以P<0.05为检验水平,发现当数据样本分为两类时,组间差异菌有50个;当分为三类时,组间差异菌有68个;当分为四类时,组间差异菌有67个;当分为五类时,组间差异菌有73个,如表2。可以看出不同的人群其差异菌的类别与其分类程度成正相关。

表2 聚类数为两至五类时组间差异菌 Tab 2 Differences in bacteria between groups when divided into two to five categories
3.4 人群分类最优模型的选择与评价
为了进一步说明可以依据差异性的肠道菌群对人群分类的可行性,基于前面试验结果将聚类后的每类样本的差异菌的定性数据分别作为模型输入指标,再分别利用NB、IBK、J48、RF四种算法进行模型的构建。并筛选出不同人群分类模型中AUC值最高的算法及分类,所建模型的应用算法及构成模型的肠道菌属性集结果见表3。

表3 聚类结果为两至五类时人群分类模型 Tab 3 Crowd classification model when the clustering results are two to five types
基于不同分类人群间差异菌所建各类模型的评价指标如表4所示,ROC曲线图见图4。所建模型的训练集和测试集,各个指标结果都趋近,这表明在应用未知数据时,所建模型不会发生故障。

图4 受试者工作特征曲线 Fig 4 Receiver operating characteristic curve

表4 聚类结果为两至五类时模型各项指标 Tab 4 Indicators of the model when the clustering results in two to five types
通过综合比较每种模型的ROC曲线图、准确性、SE、SP、BACC选择最优模型。最终,基于肠道菌群分为四类时,所建模型为最优。对于最优模型的选取,与各模型性能相关的多项评价指标,及各模型的ROC曲线图进行了分析。最优模型的性能是从训练集和测试集两个角度进行评价的。模型10折交叉验证测试的准确性在82.50%~100%,平衡精度在0.672~1.000,受试者工作特征AUC在0.842~1.000。测试集的准确性在85.29%~94.12%,平衡精度在0.808~0.904,受试者工作特征AUC在0.990~0.971。

续表2

续表3
由以上结果最终可将受试者分为四类,通过各评价指标的数据表明,本文建立的基于肠道菌群建立的受试人群分类模型有较高的可靠性,而且还说明了通过肠道菌群建立健康青年人群分类模型是可行的。对于每一类人群其优势菌群在一定程度上影响人体健康,同时宿主也会影响肠道菌群。① 第一类人群以Mitsuokai、Dolichum、Citroniae等为主要的优势菌群,高脂饮食会导致其丰度增加腹部内脏脂肪区[28],肠道中短链脂肪酸的缺乏会影响宿主健康,常常与缺乏可发酵纤维有关,因此这类人群应多食用膳食纤维,不仅可以促进有益菌代谢产生人体所需的短链脂肪酸,还可以降低这类人群的优势菌群丰度[29]。② 第二类人群主要以intestinihominis、eggerthii、onderdonkii、indistinctus等为主要优势菌群,其中intestinihominis过度扩张是微生态失衡的标志[30],其定植可降低血浆中具有抗炎作用的皮质酮的水平,从而促进肠道中的炎症应答[31];eggerthii可以合成短链脂肪酸,食用高脂低碳水使得eggerthii增多导致棕榈酸和硬脂酸等增加;onderdonkii、indistinctus菌群失调与炎症、癌症以及心理健康具有很高的相关性[32]。③ 第三类人群主要以longicatena、lactaris、johnsonii、putredinis等为主要优势菌群,longicatena属于Dorea属,是潜在等有益菌[33],在帕金森患者中其丰度降低[34],经常使用小麦、黑麦、大麦、燕麦会使其丰度会增加[35];此外lactaris具有降脂应用效果[36];Johnsonii是肠道中的有益菌,能提高血液中的还原型谷胱甘肽水平,从而改善肝脏的线粒体形态和功能,减少肝脏脂质、改善系统性的糖代谢[37];高盐饮食通过改变肠道菌群的组成及代谢,减少Putredinis,从而提升皮质酮水平以促进高血压,而Putredinis可通过其代谢产物花生四烯酸抑制皮质酮的产生[38]。④ 第四类人群以lenta、symbiosum、cateniformis为主要优势菌群,lenta被认为是溃疡性结肠炎、肝脏和肛门脓肿以及全身性菌血症的原因[39];高脂饮食会导致symbiosum丰度增加[28];经常食用土豆抗性淀粉、玉米抗性淀粉,肠道中的有益益生菌Clostridioforme显著增加,菌群产生丁酸能力更强[40]。总之,不同的人群其优势菌群不仅影响人体肠道微环境,还会间接或直接影响人体的消化、免疫以及情绪等,针对不同人群优势菌群作出不同的饮食管理、生活方式改变将有益于我们对健康管理,为个体化食疗、医疗提供新的方向。
4 总结与展望
本研究阐释了基于肠道菌群分类的健康青年人群分类模型的构建的方法及原理。基于肠道菌群种层面数据,通过聚类分析获得基于肠道菌群定性数据的健康青年人群分类,再依据分类的组间差异菌,运用数据挖掘算法构建了健康青年人群分类模型。构建模型的准确性、灵敏度、特异性、平衡精度、AUC值等几个指标验证了所建模型的可靠性。
人体的肠道微生物与宿主是共同生长的,人体不同状态呈现不同的菌群特征。曾有研究通过随机森林算法,构建了基于肠道菌群区分酒精性脂肪肝的模型[41]。这为本文通过数据挖掘的方法进行基于肠道菌群分类探讨健康青年人群分类的研究提供佐证,进一步证明了以肠道菌群分布差异作为客观指标对健康青年人群进行分类的可能性。不仅为处理微生物数据提供了一种新的策略,而且为未来深入研究肠道菌群与人体健康关系提供了参考。
