ATP7A基因新发变异致Menkes病1例
2021-12-30石晓萌路新国
石晓萌,路新国
(1.中国医科大学深圳市儿童医院风湿免疫科,广东 深圳 518038;2.深圳市儿童医院神经内科,广东 深圳 518038)
Menkes病(Menkes disease,MD)于1962年由MENKES等[1]首次报道,是一种X连锁隐性铜代谢神经退行性疾病,由于编码铜转运P型腺苷三磷酸酶(adenosine triphosphatase,ATPase)的ATP7A基因突变所致。主要表现为特征性“卷发”、进行性神经变性、难治性癫痫、结缔组织异常、新生儿低血糖、生长迟缓等。本文报道1例2020年12月我院收治的MD病例,并复习相关文献。
1 临床资料
患儿,男,6月龄,因“反复抽搐4月余、吐奶1周”入院。最初表现为颜面及口周发绀、流涎,表情呆滞,未见明显四肢动作,持续10余分钟,成簇发作,双眼向上凝视,不对称强直,左上肢屈肘强直,右上肢自然状态。3月余前加用左乙拉西坦后,抽搐仍反复发作,表现同前,持续时间可达2 h,发作18~20次/d。2月余前加用丙戊酸钠,脑电图示痫样放电,可见较多低幅尖波呈多灶发放,加用托吡酯后发作次数减少。其后加用奥卡西平及减停丙戊酸钠,发作次数增多,且出现新的发作形式,表现为舌头外伸、口唇发绀、下肢较强直,偶有上肢屈曲,持续约1~2 min,发作10~20次/d,间断右眼眨眼,伴吐奶,非喷射性,2~3次/d,治疗效果欠佳。
患儿系第2胎第2产,孕39周,顺产,出生体质量3 300 g,出生后因“窒息复苏后23 min”于外院住院,诊断“新生儿窒息,产瘤,新生儿高胆红素血症,新生儿头颅血肿,蛛网膜下腔出血”,予抗感染等对症治疗后好转。母孕期一过性高血糖,生后母乳喂养1个月,人工喂养5个月,现不能竖头、抬头,不会笑,追光、追物差。家族史、外伤及手术史、输血史、预防接种、传染病史无特殊。
体格检查:体质量7.9 kg,神志清,精神反应可。全身皮肤白,皮肤松弛,浅表淋巴结未触及肿大。头围41 cm,后枕部凹陷,卷曲发,粗硬,前囟凹陷,斜径2.0 cm×2.0 cm,张力不高,见图1。双侧瞳孔等大等圆,直径3 cm,对光反射灵敏。咽部无充血。颈软,无抵抗,未见三凹征,双肺呼吸音粗,可闻及痰鸣音,未闻及干湿啰音,心腹未见异常。关节屈曲度大,四肢肌张力低,双侧膝反射可引出。阴茎长度2 cm。
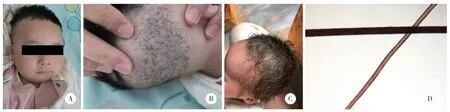
图1 患儿皮肤及毛发
辅助检查:血常规正常。尿液分析显示,细菌1 480.05/μL,结晶29.70/μL,余正常。血乳酸(lactate,Lac)1.8~6.67 mmol/L;丙戊酸35.0 μg/mL;肝肾功能、心肌酶、体液免疫正常。血气分析(静脉)pH7.562,HCO3-21.9 mmol/L,碱剩余(base excess,BE)-3.1 mmol/L,血钙 1.31 mmol/L,血糖4.8 mmol/ L,Lac值3.86 mmol/ L,铜蓝蛋白(ceruloplasmin,CER)5.8 mg/d L(正常值20~60 mg/d L);铁3.8 μmol/L(正常值9.0~32.6 μmol/L)、血 铅143 μg/L(正常值0~100 μg/L)、铜3.2 μmol/L(正常值8.79~29.54 μmol/L);尿铜9.8 μg/24 h(正常值15~60 μg/24 h)。痰培养:卡他莫拉菌(4+)。血筛查正常。尿筛查:丙戊酸、2-丙基-3羟基戊酸、2-丙基-5羟基戊酸增高,可能与使用抗癫痫药物有关。泌尿系统超声显示:右肾高回声团块,考虑结石。胸部X线片:双侧肋骨末端膨大。上消化道超声未见异常。心电图示窦性心律,大致正常心电图。颅脑磁共振(5月龄)显示:双侧大脑半球多发软化灶伴胶质增生,双侧额顶颞部硬膜下积液/积血,脑萎缩,幕上脑室扩张,见图2。脑电图(6月龄)显示:各导大量弥漫性慢波,双侧枕区为主大量不规则慢波混合棘波、尖波及低波幅快活动连续发放,可波及双侧后颞区。

图2 颅脑磁共振图像(5月龄)
诊治经过:入院后停用奥卡西平,继续口服左乙拉西坦38 mg/(kg·d)及托吡酯片抗癫痫治疗,并将托吡酯剂量由2 mg/(kg·d)上调至3.2 mg/(kg·d)。辅以头孢曲松50 mg/(kg·d)抗感染,吸痰、雾化、补液等对症治疗后,Lac值降至2.94 mmol/L,但仍有每日抽搐发作,家属要求出院。
基因检测:根据患儿病情需要,家长签署知情同意书,完善全外显子基因检测。结果显示,X染色体ATP7A基因5号外显子存在半合子变异:c.1435C>T(p.Gln479Ter),见图3。该变异为无义突变,在参考人群基因频率数据库(The Genome Aggregation Database,gnomAD)中最小等位基因频率未见记录,该变异被Clinvar数据库报道为Pathogenic(http://www.ncbi.nlm.gov/clinvar/variation/618540)。经家系验证后提示受检者父亲未见突变,受检者母亲杂合突变,此基因突变导致第479号氨基酸由谷氨酰胺变为终止密码子(p.G1n479Ter)。根据美国医学遗传学与基因组学学会(The American College of Medical Genetics and Genomics,ACMG)指南,该变异为功能丧失突变(loss-of-function,LOF),该突变位点在ESP数据库、千人数据库、EXAC数据库中未见,为新发突变,截断突变c.1435C>T导致转录提前终止,该变异为可能致病变异。
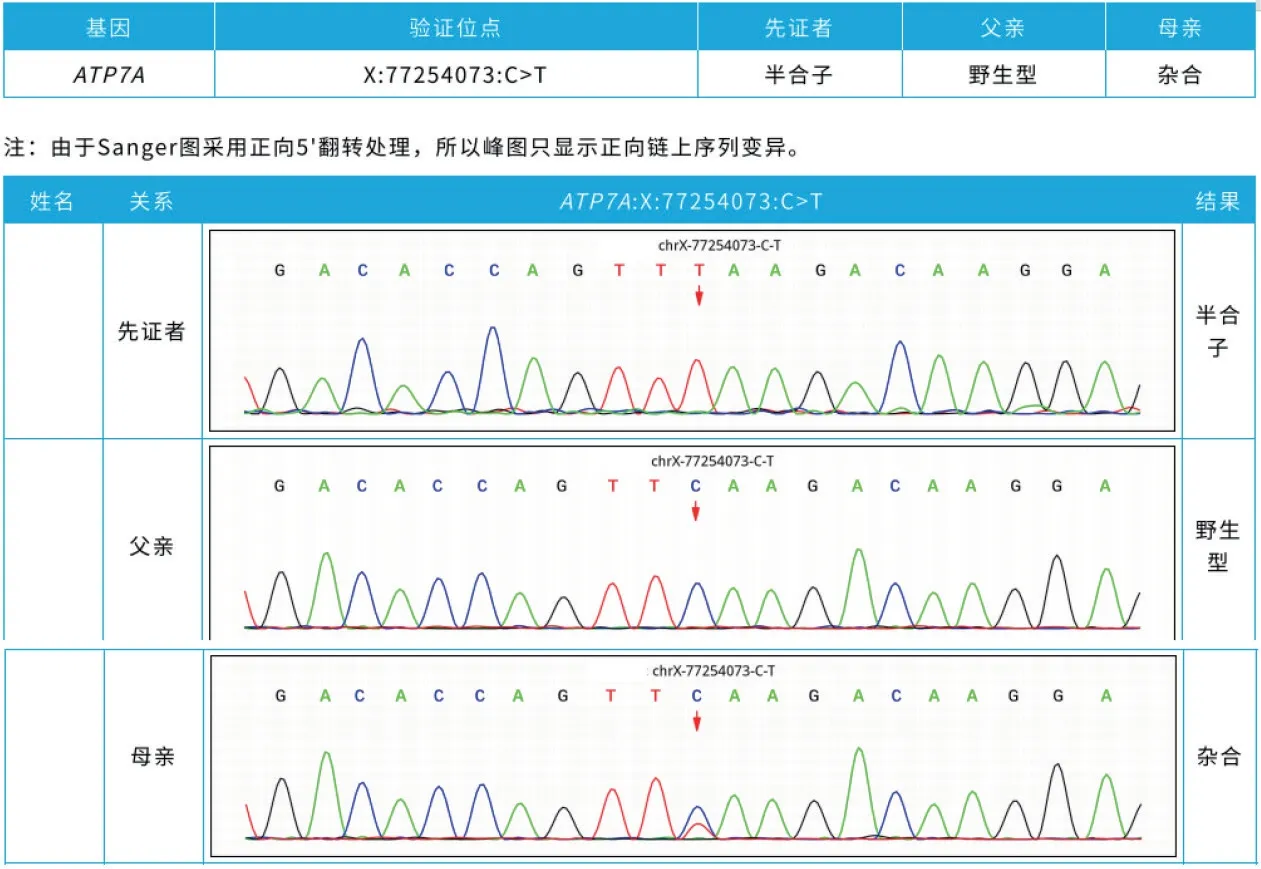
图3 ATP7A基因测序结果显示患儿存在c.1435C>T位点变异,患儿母亲为携带者,父亲无该位点变异
结合患儿临床特征、实验室检查及基因结果,患儿出院诊断为“MD,难治性癫痫(局灶性发作),癫痫性脑病,全面发育迟缓,急性支气管炎,右肾结石,高铅血症”。
2 讨论
MD是一种由位于Xq21.1的ATP7A基因突变引起的X染色体隐性遗传的进行性全身性铜代谢疾病,1962年由MENKES等[1]首次报道。发病率约为1/300 000~1/100 000,澳大利亚可能高达1/40 000,男性多见[2-3]。ATP7A基因包含23个外显子,分布在约150 kb的基因组DNA中,编码位于跨高尔基网络(trans-Golgi network,TGN)的1 500个氨基酸残基的铜转运P型ATPase,具有6个氨基末端铜结合位点和1个具多个功能结构域的催化转导核心;在肠细胞、胎盘、构成血脑屏障的星形胶质细胞、脑血管内皮细胞及神经元和脉络丛细胞中表达[4]。根据残留的ATP7A活性不同,MD可分为经典MD(占90%以上)、枕角综合征(occipital horn syndrome,OHS)和X连锁远端脊肌萎缩症-3(X-linked distal spinal muscular atrophy type 3,SMAX3)。
关于MD的致病机制,一种广泛被认可的观点是人肠细胞外的Cu2+被二价金属转运蛋白1(the divalent metal transporter 1,DMT1)结合并还原为Cu+,通过肠上皮细胞顶膜上的铜转运蛋白1(copper transporter 1,CTR1),再经超氧化物歧化酶的铜伴侣(copper chaperone for superoxide dismutase,CCS)、细胞色素C氧化酶铜伴侣蛋白(cytochrome C oxidase copper chaperone,COX17)和抗氧化蛋白(antioxidant protein 1,ATOX1)将Cu+靶向转至超氧化物歧化酶(superoxide dismutase,SOD1)/细胞色素C氧化酶(cytochrome c oxidase,CcO)/高尔基体途径,ATP7A接受ATOX1中的Cu+,介导铜从肠细胞外流到血液中,或经ATOX1/ATP7B/CER途径分泌到胆汁或血液中,正常血清铜浓度约为109 μg/100 mL,其中90%与CER结合,其余的铜离子与白蛋白或游离氨基酸结合。遗传小鼠模型已经证明,ATOX1/ATP7A途径的缺陷会导致铜分布的缺陷,引起相应的病理变化[4]。ATP7A、ATP7B负责调节铜离子浓度,并转入如酪氨酸酶、多巴胺单加氧酶、CcO、赖氨酰氧化酶、CER等10余种铜依赖蛋白酶;CER具有铜依赖氧化酶活性,能将Fe2+转化为Fe3+,对转铁蛋白介导的血浆中铁的转运有重要意义[5]。本患儿血清铁3.8 μmol/L,考虑为CER降低影响铁代谢所致。
以“Menkes病”为关键词,检索中国知网(http://www.cnki.net)及万方数据库(https://www.wanfangdata.com.cn)建库至2021年2月,选取其中14篇国内文献[6-19]共43例临床信息相对完整的患者资料,加上本例患者,共44例MD患者。44例(100%)均为男性,平均中位发病年龄3个月(3 d至7个月)。33例有首发症状的患者中,以抽搐、发育迟缓为首发症状者分别占81.8%(27/33)、18.2%(6/33)。44例患儿中,癫痫发作40例(91.0%),精神运动发育迟缓44例(100%),皮肤白皙44例(100%),头发黄、卷曲、稀疏42例(95.5%),肌张力降低36例(81.8%),漏斗胸7例(15.9%),腹股沟疝8例(18.9%),颅内出血2例(4.5%),膀胱憩室1例(2.3%)。头颅磁共振显示血管迂曲样改变(4/4)、基底节异常信号(2/7)、脑发育不良(2/7)、脑萎缩(3/7)、硬膜下积液(1/7),19例患者血清CER为低水平;32例患者中发现25种ATP7A突变,其中突变位点c.3914A>G(p.D1305G)3例,2~12外显子大片段缺失3例,余均为个例。33例患者中,已知突变类型包括9个大片段缺失、9个错义突变、6个剪切突变、5个无义突变、2个缺失/插入突变、2个小片段缺失。可以推断中国MD患儿中,c.3914A>G(p.D1305G)、2~12外显子大片段缺失可能为热点突变,突变类型以大片段缺失及错义突变为主,而FUJISAWA等[20]报道的66例日本MD患者中无义突变占多数(24%)。至今已报道了MD约379种不同的突变(http://www.hgmd.cf.ac.uk/ac/index.php)。本例患儿ATP7A基因变异点位于5号外显子,第479号氨基酸由谷氨酰胺变成终止密码子(p.Gln479Te),为功能丧失突变,该无义突变既往未见报道。患儿母亲为携带者,本例患儿的基因突变为国内外首次报道,丰富了MD的遗传基因谱。
MD影响的器官包括脑、肺、胃肠道、尿路、结缔组织和皮肤,临床特征包括抽搐、精神运动发育迟缓、视神经萎缩、喂养困难、自主神经症状(体温过低、低眼压)、呼吸暂停、感染、特殊面容、结缔组织症状(卷发、色素减退、膀胱憩室、关节和皮肤松弛),其中结缔组织症状为主要表现,光镜下发现的毛发异常包括卷毛(轴上180°扭曲)、丝状体和结节毛发[5,21];本例患儿新生儿期高胆红素血症,以早发性癫痫起病,精神运动发育迟缓,皮肤白皙、松弛,漏斗胸,关节屈曲度大,肌张力减弱,多种抗癫痫药物口服治疗效果均欠佳,进行性脑萎缩,CER及血清铜降低,均与既往报道特征相符,结合基因检测结果,属经典MD表型。患儿毛发新生儿期呈稀疏、金黄色,40日龄时转为黑色、粗硬,表现与典型MD稀疏头发有差别,伴随呕吐等胃食管反流症状,提示不同表型可能与不同ATP7A基因的遗传异质性有关。由于血管结缔组织异常和进行性脑萎缩,硬膜下血肿在MD中很常见,本例患儿新生儿期有蛛网膜下腔出血、头皮血肿,虽无颅脑血管磁共振证实,但仍提示患儿可能有颅内血管迂曲畸形。杨广娥等[16]曾报道过1例以新生儿期颅内出血为首发症状的MD,因此,儿科临床医生必须提高对MD血管改变的认识,保持警惕,防止误诊和不必要的非意外损伤怀疑。
目前主要应用组氨酸铜治疗MD。回顾性研究[22]证实,有效的组氨酸铜治疗方案为第1年2次/d皮下注射250 μg组氨酸铜,以后改为1次/d皮下注射250 μg,期间监测血清铜和CER水平。对于ATP7A突变中铜转运能力有一定程度保留的患者,铜替代治疗可能对神经系统功能有改善。KALER等[23]报道了1例ATP7A无义突变(R201X)患者生后8 d接受组氨酸铜皮下注射,治疗效果良好,基本无神经认知缺陷,支持了早期诊断和治疗的重要性。MD预后不良,患者多于6个月至3岁内死亡,多并发顽固性癫痫、硬膜下血肿、失明、反复感染,死因常与血管并发症或呼吸道感染有关。因此,遗传咨询及产前诊断对于致死性疾病MD的预后极为重要。
