NixP(tx=1-9)合金团簇结构稳定性与磁性的第一性原理研究
2021-12-30岳莉
岳 莉
(凯里学院,贵州凯里 556011)
合金团簇与纯金属团簇相比,电子结构更加复杂,其性质与团簇的大小和化学成分有关[1].镍,是一种硬而有延展性并具有铁磁性的金属,它能够高度磨光和抗腐蚀,在军工、航空航天、机械、不锈钢等领域有着重要应用,它的化学性质较活泼,主要用于合金及催化剂,对Ni 团簇和以Ni 为基体的合金团簇研究已有许多[2−4].由于过渡金属原子中含有d 电子,形成的合金团簇就具有更为特殊的电子结构和物理化学性质[5].铂是过渡金属,有良好的延展性、导热性和导电性,化学性质不活泼,有很高的化学稳定性和催化活性,对以Pt 为基体的合金团簇研究也有不少[5−6],而合金团簇的物理化学性质是可以通过团簇的组成、尺寸和化学顺序进行调节的,目前,对NixPt团簇结构稳定性与磁性的研究还未见报道.为此,本文采用基于密度泛函理论的第一性原理方法,对NixPt(x=1−9)团簇结构稳定性与磁性进行研究.
1 计算方法
NixPt(x=1−9)合金团簇构型设计时,对于总原子数N=x+1 ≤8 的团簇,几乎考虑到每个尺寸的所有构型,同时参考Nix+1团簇构型.由于团簇结构的简并电子态会对其稳定性造成影响,因此,对经筛选后的NixPt稳定结构设置不同自旋多重度进行优化,再比较NixPt团簇总能量,能量最低的构型即为NixPt 基态结构.设计NixPt(x=1−9)团簇结构的具体方法如下:首先在稳定、次稳定、亚稳定Nix+1和同族同尺寸团簇非对称位置上用一个铂原子替代一个镍原子;接着在稳定、次稳定、亚稳定Nix团簇的不同位置上添加一个铂原子;最后在Nix-1Pt 团簇非对称位置上添加一个镍原子.同时,为了排除部分局域最小值而更加接近全局最小值,在对每个初始结构优化的过程中均没有考虑对称性.
为了能更精确地描述积分和电子自旋的贡献,计算过程中考虑自旋非限制,角动量多级扩展使用十六偶极机制和双数值极化基组.本文采用广义梯度(GGA)近似下的BLPY 关联函数,在总能、原子位移和能量梯度的收敛精度设为1×10−6eV、5×10−4Å 和1×10−5eV/Å下进行计算.
2 结果与分析
2.1 团簇的最低能量结构
图1和2分别显示了Nix+1(x=1−9)团簇和NixPt(x=1−9)合金团簇的最低能量结构,其中黑色小球代表Pt 原子,表1、表2分别列出了Nix+1(x=1−9)团簇和NixPt(x=1−9)合金团簇的计算结果.
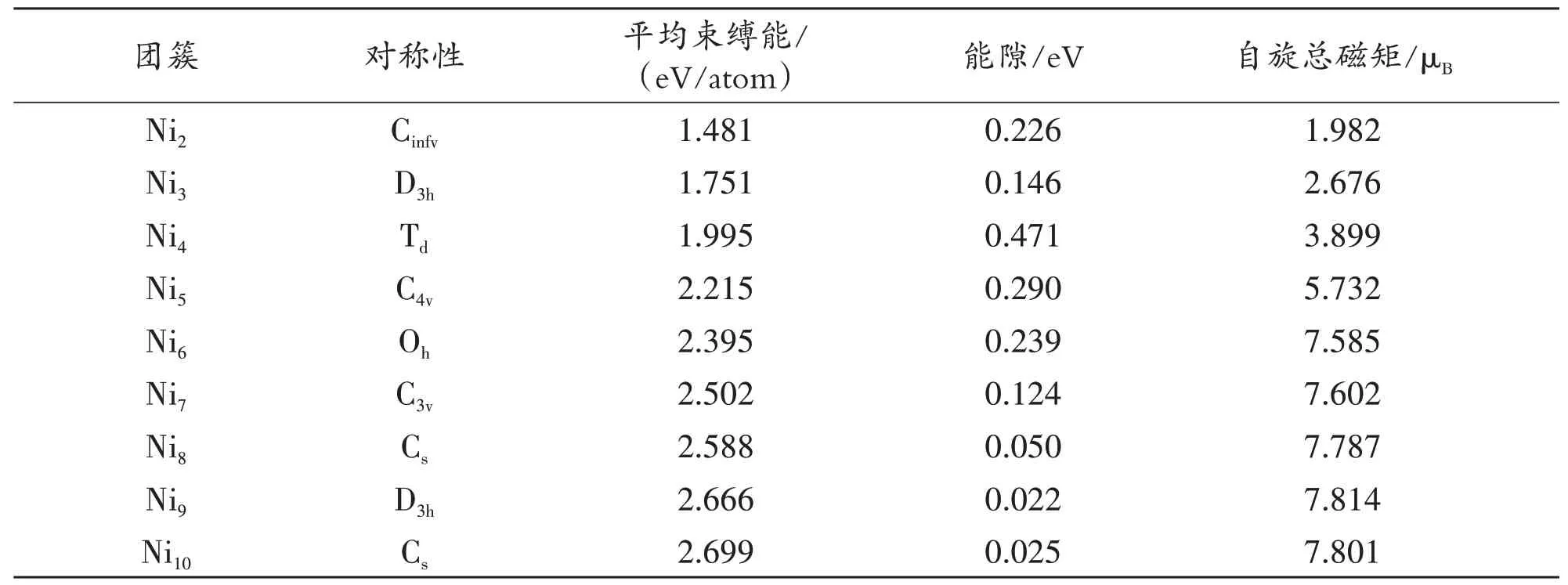
表1 Nix+1(x=1-9)团簇基态结构的对称性、平均束缚能、HOMO-LUMO能隙、自旋总磁矩

表2 NixPt(x=1-9)团簇基态结构的对称性、平均束缚能、能隙、铂原子电荷、自旋总磁矩

图1 Nix+1(x=1−9)团簇最低能量结构
从图1 的Nix+1(x=1−9)和图2 的NixPt(x=1−9)团簇最低能量结构可以看出,Ni6Pt、Ni7Pt 分别与Ni7、Ni8不仅形状没变而且对称性也没变,说明对于这些尺寸,Pt原子的掺杂并没有改变纯Ni团簇几何构型.另外,NiPt、Ni2Pt、Ni3Pt 分别与Ni2、Ni3、Ni4虽然有相似的最低能量结构形状,但有的对称性却发生了变化,如纯Ni3金属团簇的最低能量结构是等边三角形而合金Ni2Pt 团簇的最低能量结构却是等腰三角形,纯Ni4金属团簇的最低能量结构是各边均相等的三棱锥,合金Ni3Pt团簇的最低能量结构却是底为等边三角形的三棱锥,合金Ni4Pt、Ni5Pt、Ni9Pt团簇的最低能量结构与纯Ni5、Ni6、Ni10团簇的最低能量结构形状和对称性都不一样,如纯Ni5团簇是底为四边形的三棱锥而合金Ni4Pt团簇却是共等边三角形底的两个三棱锥.

图2 NixPt(x=1−9)团簇最低能量结构
2.2 团簇的能级与稳定性
团簇的平均束缚能和HOMO−LUMO 能隙是反映其稳定性的主要依据,平均束缚能越大结构越稳定,HOMO−LUMO 能隙越大,团簇化学性子越稳定.为了描述稳定性,作者计算了团簇的平均束缚能和HOMO−LUMO能隙,并将其绘制于图3和图4中.

图3 团簇平均结合能随尺寸增加的变化规律

图4 团簇能隙随尺寸增加的变化规律
从图3 的团簇平均结合能随尺寸增加的变化规律可以看出,NixPt(x=1−9)团簇的平均束缚能随着原子总数N=x+1的增多而增大,说明NixPt(x=1−9)团簇的稳定性随尺寸增加而加强.与纯Nix+1(x=1−9)团簇的平均束缚能相比较看出,合金NixPt团簇的平均束缚能与纯Nix+1团簇的走势大致相同,都随着原子总数的增多而增大,但合金NixPt 团簇的平均束缚能都比相同原子总数的纯Nix+1团簇的高,说明Pt原子的掺杂增加了团簇的稳定性,其原因是原子的配位数随原子总数增加而增多,合金NixPt团簇大于纯Nix+1团簇,使得合金NixPt团簇结构总能量降低较多,释放的能量较多,合金团簇的稳定性稍高于同尺寸的纯团簇.
从图4 的团簇能隙随尺寸增加的变化规律可以看出,NixPt(x=1−9)团簇的HOMO−LUMO能隙随着原子总数N=x+1的增多在振荡过程有增大有减小.HOMO−LUMO能隙直接影响着团簇的化学活性和稳定性,它的大小反映了电子从占据轨道向空轨道跃迁的能力,代表了参与化学反应的能力,HOMO−LUMO 能隙值较大时,电子不容易发生跃迁,则被认为具有较强的化学稳定性,HOMO−LUMO 能隙值较小时,电子跃迁能力强,则被认为具有较强的化学活性.与纯Nix+1(x=1−9)团簇的HOMO−LUMO 能隙相比较看出,合金NixPt团簇的HOMO−LUMO 能隙与纯Nix团簇的走势不一致,但在N=4 时都有最大值,说明合金Ni3Pt 团簇与纯Ni4团簇一样有较强的化学稳定性,合金Ni9Pt 团簇比其周围合金团簇的化学稳定性强,这与具有相同原子总数的纯Ni10团簇相反.
2.3 团簇的磁性
团簇的磁矩是宏观之下磁力的来源,为此作者计算了团簇的自旋总磁矩、铂原子电荷和铂原子自旋磁矩.
从图5 的团簇自旋总磁矩随尺寸增加的变化规律可以看出,NixPt(x=1−9)团簇的自旋总磁矩随着原子总数N=x+1 的增多迅速增大,最后趋近饱和值7.792µB.与纯NixPt(x=1−9)团簇的自旋总磁矩相比较看出,合金NixPt 团簇的自旋总磁矩与纯Nix+1团簇的走势相似,合金NixPt 团簇的自旋总磁矩总体比纯Nix+1团簇的小,但随着原子总数N的增多最后趋于相等,且纯Nix+1团簇的自旋总磁矩在N=6时达到饱和,而合金NixPt团簇是在N=7才达到饱和.

图5 团簇自旋总磁矩随尺寸增加的变化规律
从图6 的Pt 原子电荷与自旋磁矩随尺寸增加的变化规律可以看出,随着NixPt(x=1−9)团簇原子总数N=x+1 的增多,Pt 原子获得电子逐渐增加,Pt 原子自旋磁矩在振荡过程中急剧减小,说明随着原子总数N的增多,NixPt团簇中用于成键的电子逐渐增多,当N=5、10 时,Pt原子获得电子达到较大值、Pt 原子自旋磁矩有较小值,表明Ni4Pt、Ni9Pt 团簇的稳定性较好,这与平均束缚能部分分析的结果吻合的很好.
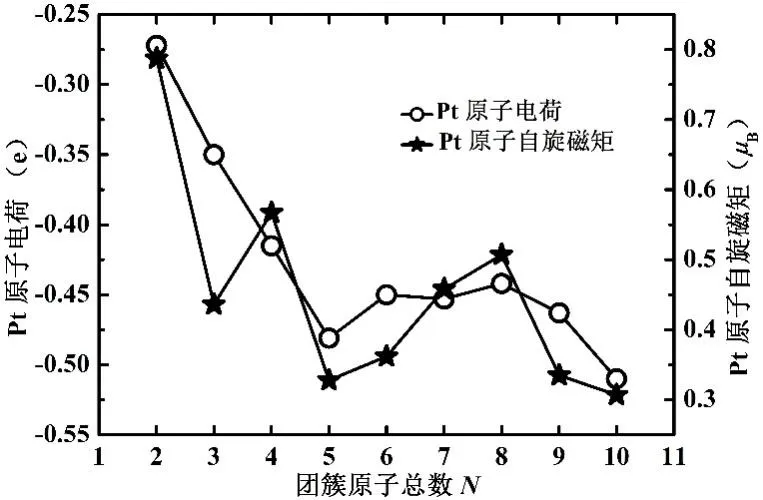
图6 Pt原子电荷与自旋磁矩随尺寸增加的变化规律
3 结论
利用广义梯度(GGA)近似下的BLYP关联函数对NixPt(x=1−9)团簇及纯NixPt(x=1−9)团簇的结构稳定性与磁性进行系统的研究,得到以下结论:随着团簇原子总数N的增加Pt原子的掺杂对纯Ni团簇的几何结构改变不大,但是,平均束缚能却改变许多,相对于纯团簇而言,Pt原子的掺杂使所有研究尺寸团簇的稳定性都得到增强;NixPt(x=1−9)团簇的HOMO−LUMO 能隙随着原子总数N=x+1的增多出现振荡行为;随着团簇总原子数的增加,合金NixPt团簇的自旋总磁矩总体比纯Nix+1团簇的小,但最后都趋于相近,而纯Nix+1团簇的自旋总磁矩在N=6时达到饱和,而合金NixPt团簇是在N=7才达到饱和,整个过程中,Pt原子得到的电子数逐渐增大.
