X-连锁淋巴细胞异常增生症1例
2021-12-22林丽曼柯创宏黄宇戈胡海玲广东医科大学附属医院儿童医学中心广东湛江54000广东省吴川市人民医院儿科广东吴川54500
林丽曼,柯创宏,黄宇戈,胡海玲,王 倩 (.广东医科大学附属医院儿童医学中心,广东湛江 54000;.广东省吴川市人民医院儿科,广东吴川 54500)
X-连锁淋巴细胞异常增生症(XLP)又称为Dun‐can 病,是一种罕见X 连锁原发性免疫缺陷疾病。Purtilo 等[1]在邓肯家族中首次发现,根据基因和蛋白质改变类型XLP 分为1 型(XLP-1)及2 型(XLP-2)[2]。XLP-1 型是因编 码SH2 结构域蛋白1A 的SH2D1A基因突变,引起编码蛋白异常,导致T淋巴细胞和NK细胞的功能受损,在感染等因素下可导致噬血细胞性淋巴组织细胞增生症(HLH)[2];而XLP-2 型是由BIRC4基因突变导致其编码的XIAP 蛋白异常表达而发病[3]。目前国内有关XLP-1 型报道极少,现分享1例经基因检测为XLP-1型及其家系基因,旨在提高临床医生对该病的认识。
1 病例资料
患儿,男,3 岁,2017 年5 月25 日因“发热13 d,抽搐1 次”入院。患儿无明显诱因发热,呈稽留热,伴抽搐1 次,呈全身痉挛性大发作,发作持续约数分钟后缓解。家族史:父母和妹妹健在,其母亲胞姐2 个孩子因“肝大”治疗无效死亡(具体不祥)。入院查体检:体温39 ℃,精神疲倦,颈部可触及多个肿大的淋巴结,最大约2.0 cm×1.5 cm×1.5 cm,质中,皮温正常,无压痛,且有多个淋巴结融合,腹稍膨隆,肝肋下4 cm可触及,质中,脾脏肋下未触及。
辅助检查:血白细胞(WBC) 11.47×109/L,单核细胞21.28%,中性粒细胞0.9×109/L,Hb 86 g/L,PLT 166×109/L,EBV-IgM 2.55(阳性),纤维蛋白原(FIB)1.51 g/L,活化部分凝血活酶时间(APTT)97.3 s,丙氨酸氨基转移酶916 IU/L,谷草转氨酶2 106.8 IU/L,白蛋白21.5 g/L,总胆固醇1.65 mmol/L,自然杀伤细胞CD3-/CD16+CD56+7.18%,血氨328.1 μmol/L,铁蛋白9 835 μg/L。外周血细胞涂片(图1a):异型淋巴明显增多,占21%。骨髓细胞学涂片(图1b):骨髓增生活跃,粒系增生明显活跃,红系增生减低,淋巴系增生,可见异型淋巴占5%,巨核系增生,噬血细胞占1%,吞噬红系、粒系及血小板。腹部及颈部B 超提示:肝大、脾大,副脾声像图,双侧颈部及锁骨上、下窝淋巴结肿大,内部结构欠规则。
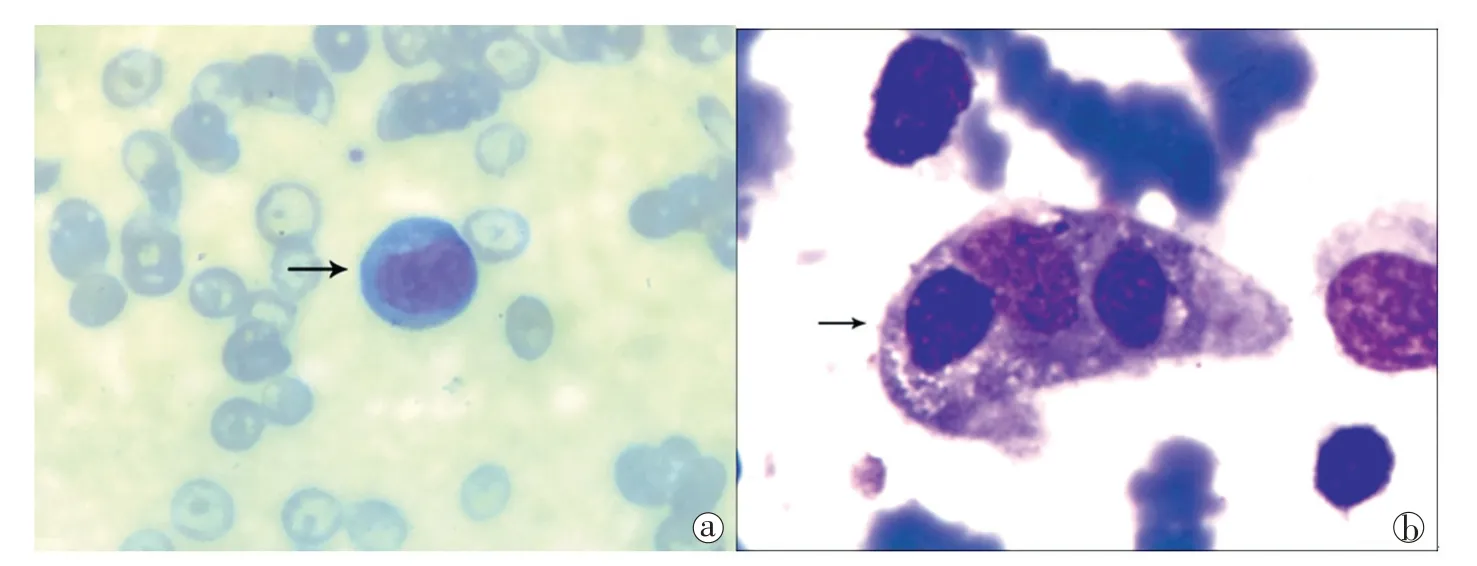
图1 外周血细胞涂片/骨髓细胞血涂片
治疗经过:入院初步诊断为传染性单核细胞增多症,予更昔洛韦抗病毒、热毒宁联合地塞米松治疗后仍高热,凝血4 项异常,血常规示三系下降[WBC(11.47~6.89) ×109/L,PLT(166~81) ×109/L,Hb(86.0~73.9 g/L)],肝功能受损,治疗效果不佳。为进一步诊断,予骨髓细胞学检查可见噬血现象。结合患儿高热、血细胞减少、低FIB、铁蛋白升高、脾大、骨髓细胞见噬血现象,满足HLH-2004诊断标准8条指标中的5条,诊断为噬血细胞综合征。同时患儿出现谵妄,进展为浅昏迷,腹部进行性膨隆,大理石花纹,凝血功能障碍,血气分析:pH 7.28,PO2109 mmHg,PCO213.5 mmHg,BE -19.5 mmol/L,AG 34.6 mmol/L,Lac 15.6 mmol/L,Ca 0.98 mmol/L,提示代谢性酸中毒合并电解质紊乱等,予升级抗生素为美罗培南,丙种球蛋白免疫支持,甘露醇降颅压,输注血浆、血小板,多巴胺联合山莨菪碱改善循环,乳果糖降血氨,纠正电解质紊乱等治疗后病情仍继续加重,有多器官功能衰竭征象,告知家属预后极差,拟HLH-2004 化疗以及完善遗传病医学外显子组基因测序,家长放弃治疗,出院后死亡。
基因检测:2017-7-19 遗传病医学外显子组基因测序示检测到受检者携带ACAD9(NM_014049.4)基因一个杂合致病突变,高通量检测结果提示受检者存在SH2D1A基因的半合子缺失。ACAD9(NM_014049.4)基因突变位点为:c.1579C>T(Exon16),p.(Gln527*),该突变为无义突变,既往ESP、千人基因组以及db‐SNP147 数据库均未见收录,经MutationTaster 预测的突变评级为可疑致病(ACAD9基因如发生致病突变常以常染色体隐性方式遗传,患者的父母往往均携带致病突变,携带一个杂合致病突变不会发展成为患者)。其母亲、二姨妈测序结果与患儿的突变位点一致,同样检测到ACAD9(NM_014049.4)基因突变和SH2D1A基因的杂合缺失,三姨妈检测到SH2D1A基因的杂合缺失。患儿父亲、其胞妹及其二姨妈的两个女儿均未检测到ACAD9(NM_014049.4)和SH2D1A基因的半合子缺失(如图2)。
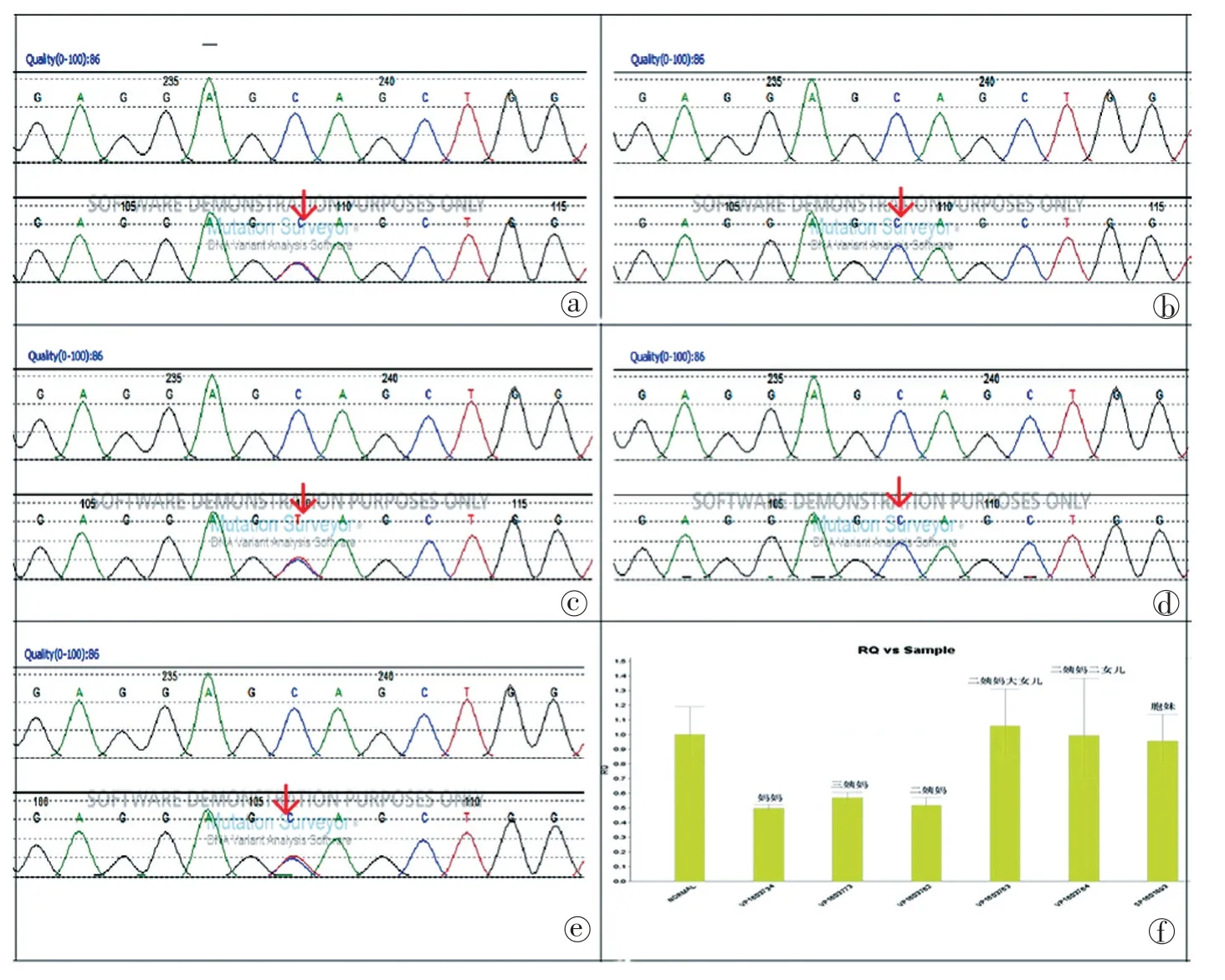
图2 Sanger测序和QPCR结果
2 讨论
2.1 遗传模式
XLP-1 型的致病基因是位于X 染色体长臂(Xq25),是由编码信号淋巴细胞激活分子(SLAM)相关蛋白(SAP)的SH2D1A基因失活突变引起的,约60%~70%的XLP基因突变是由编码SAP的基因突变引起,SAP 调节着免疫细胞功能、造血细胞相互作用的功能等,其突变导致SAP 功能丧失[3-4]。SH2D1A基因位于X染色体上,这可以解释为什么母亲没有明显的临床表现,因此绝大多数携带XLP 基因(编码SAP)杂合突变的女性在临床上不受影响,而只有男性发病[5-6]。故XLP-1 发病特征是男性发病,女性为携带者。本研究取得了母系的基因分析样本,检测母系其他亲属是否存在基因突变,从而验证X连锁隐性遗传疾病的遗传规律,即女性为基因突变携带者。该患儿X染色体上的SH2D1A基因发生缺失,其母亲、两个姨妈均为携带者基因型,其父、其妹为正常基因型。根据患儿典型的临床表型与基因型,诊断XLP-1型明确;患儿的母系检测结果符合XLP-1 型典型遗传规律,其家谱图如下(图3)。

图3 母系家族XLP系谱
2.2 临床表型与基因型的相关性
在XLP-1 型中,SH2D1A基因型与临床表型之间没有一致的关联,基因型相同的家族中其临床表型有很大的差异,缺乏基因型-临床表型相关性[7],其临床表型是:暴发性传染性单核细胞增多症(FIM)(60%)、异常丙种球蛋白血症(30%)和淋巴瘤(30%)等[8]。同时也有文献指出:XLP-1 型的主要表型是HLH[7,9]。而PHL突变的基因位点也包括XLP的致病基因在内[10]。这些表型可单独出现,也可几种同时或先后出现,并有可能从一种表型进展到另一种表型[7,11]。FIM/HLH 是XLP 最致命的并发症,其中FIM 典型临床表现为发热、淋巴结肿大、广泛的组织损伤,尤其是肝脾等脏器,其中肝脏、骨髓为最易受累器官,伴有神经系统受累表现,如抽搐、意识障碍等,且FIM 可发展为HLH,即骨髓检查可见噬血细胞,因此FIM/HLH 是XLP最严重、预后最差的临床表型[8]。
本病例为男性,以FIM、HLH 临床表型出现,其HLH可能是继发于FIM。在疾病过程中,出现全血细胞减少、肝衰竭和神经系统疾病损伤,结合遗传病医学外显子组基因测序诊断为XLP-1型,也经Sanger测序验证患儿母亲、二姨妈及三姨妈为SH2D1A基因型的携带者,与XLP-1 型临床表型符合,同时也符合PHL的诊断。
2.3 诊断和预后
诊断该病的客观标准包括临床表型、相关家族史及基因型,对于男性患儿出现FIM、HLH、淋巴瘤及低丙种球蛋白血症等应高度警惕XLP-1型,尽早完善相关基因型协助诊断。XLP-1 型有严重发病率和高死亡率,根据文献报道:XLP-1 型的预后有了很大改善,与不同临床表型相关的死亡率发生了变化,与HLH相关的死亡率下降到65%,淋巴增生性疾病的死亡率下降到8%,丙种球蛋白血症下降到5%。目前,异基因造血干细胞移植是唯一可用于XLP-1 型的确定性治疗方法[7,12-13]。
2.4 体会
本研究报道了XLP-1型临床表型和基因型,符合X 连锁隐性遗传规律。从本例疾病发生、发展过程可得出早期诊断对该病不同转归的重要意义。然而XLP在临床表现上具有异质性,在没有可疑家族史的情况下,早期识别可能是困难的。此外,本病具有遗传倾向,如果患儿的父母或母系家属需继续生育,应进行产前诊断。另外,儿科医师应提高对该病的认识,对有反复高热的、有HLH 临床表现或阳性家族史的患儿,应高度警惕本病。
