CpG ODN 佐剂的研究进展及其药学研究的思考
2021-12-21徐莉李敏
徐莉,李敏
国家药品监督管理局药品审评中心,北京100022
目前国外已有应用CpG ODN 作为佐剂的预防性疫苗上市,且多个疫苗处于临床试验研究阶段,国内也有疫苗正在开展临床试验研究,成为目前疫苗研发的热点之一。本文拟对CpG ODN 佐剂的相关研究进展作一报道,并从技术审评角度对其药学研究提出一些思考。
1 CpG ODN 佐剂的概述
CpG ODN(CpG 基序的寡核苷酸,以下简称CpG)中文名称为非甲基化胞嘧啶和鸟嘌呤二核苷酸为核心的寡聚脱氧核苷酸,是人工合成的18 ~ 30 bp 的具有免疫刺激活性的非甲基化胞嘧啶-鸟嘌呤二核苷酸的DNA 重复序列,其中为提高其稳定性并延长其体内半衰期,部分或全部磷酸二酯键被硫代磷酸二酯键取代。CpG 根据其结构特征,可不同程度诱导细胞免疫和体液免疫,增强机体免疫应答。
CpG 的早期研究源于对癌症的治疗。TOKUNAGA 等[1]通过对牛减毒分枝杆菌提取结核素,证实了细菌DNA 具有抗肿瘤和提高NK 活性的功能,能够诱导Ⅰ、Ⅱ型干扰素的产生。随后的研究发现,非甲基化的CpG DNA(即CpG 基序)也具有免疫刺激作用[2]。非甲基化CpG 广泛存在于原核生物如细菌基因组中,人和脊椎动物的CpG 绝大多数被甲基化,仅有非甲基化的CpG 具有免疫刺激效应。
人B 细胞及pDC 细胞是表达TLR9 并直接对CpG 刺激产生应答的细胞。CpG 与TLR9 结合后,通过MYD88、IRAK 和TRAF6 最终激活多种转录因子,包括 NF-κB、AP1、CEBP 和 CREB。这些转录因子直接上调细胞因子和趋化因子基因表达。CpG 通过激活这些细胞启动免疫刺激,最终导致自然杀伤细胞(NK)间接成熟、分化和增殖的级联反应,T 细胞和单核细胞/ 巨噬细胞一起分泌细胞因子和趋化因子,产生促炎症因子(IL-1、IL-6、IL-18 及 TNF)和Th1偏向性免疫(IFNγ、IL-12)[3]。通过上调 pDC 细胞CD80、CD86、CD40 和 MHC 分子的表达,增加抗原处理/提呈和CD8+T 细胞反应,驱动Th1 型免疫和CD8+CTL 细胞毒性。而TLR9 依赖的B 细胞激活导致抗原特异性体液反应增加和IgG 类别转换。CpG-DNATLR9-介导的细胞信号转导通路见图1。CpG 促进的先天免疫与适应性免疫应答免疫机制见图2。
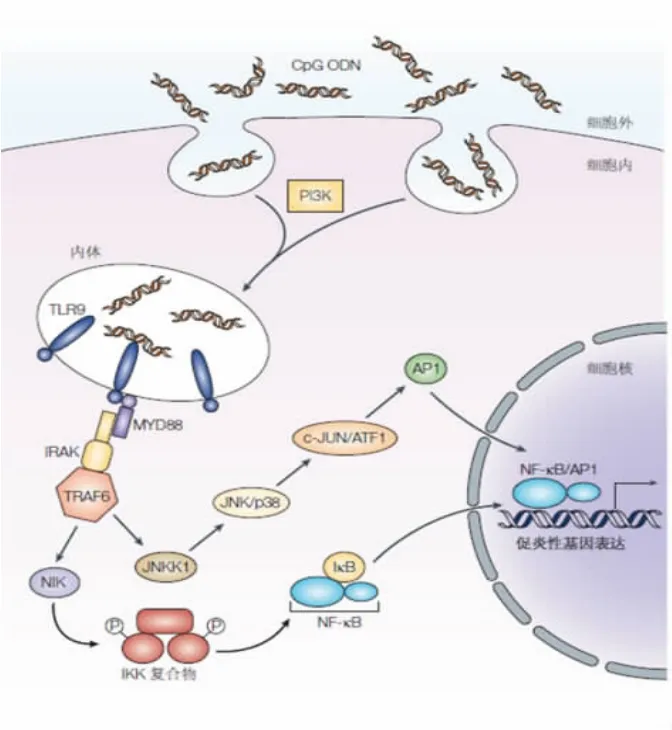
图1 CpG-DNA-TLR9-介导的细胞信号转导通路

图2 CpG 促进的先天免疫与适应性免疫应答免疫机制
根据结构、生物学性质以及体外活化免疫细胞情况,可将CpG 分为3 个型别。在硫代磷酸酯主链上的多个CpG 基序被归类为K 型ODN(也称为B型),是B 细胞活化、pDC 细胞和单核细胞成熟的强诱导因子。D 型ODN(也称为A 型)通常含有单一以天然磷酸二酯键链接的CpG 基序,主链为磷酸二酯 / 硫代磷酸二酯混合骨架,基序两侧有回文序列及 3′和 5′-末端的 poly-G 尾,这种结构特征可导致CpG 分子在溶液中形成复杂多聚体。该型CpG 可激活NK 细胞,促进pDC 细胞成熟并分泌IFNα,但对B 细胞无影响。第3 类C 型 ODN 在核苷酸单元上与K 型相似,完全由硫代磷酸核苷酸组成,结构上含有与D 型CpG 类似的回文CpG 基序,因此可形成茎环结构或二聚体。C 型ODN 可诱导B 细胞和 pDC 细胞活化及 IFNα 产生[4]。K 型 ODN 对 DNA酶的敏感性相对低,可在动物体内存留更长时间而发挥持久作用[5]。不同CpG 基序作用于不同物种引起的免疫刺激效应有所差异,具有种属特异性,同时CpG 的核心序列、回文结构等均对其活性有显著影响[6]。其主要原因可能是不同种属的TLR9 受体蛋白结构不相同。
CpG 可通过改善抗原递呈细胞的抗原摄取,激活抗原递呈细胞的功能性成熟,产生细胞因子和趋化因子从而增加抗原的免疫原性。CpG 具有稳定、低成本、易合成、高效低毒等优点[7],具有诱导Th1免疫反应的偏好性,并可延长产生抗体的持久性[8],是疫苗研究中应用广泛的佐剂。
2 CpG ODN 佐剂的临床应用情况
目前国内外已获批上市或处于临床试验阶段的CpG 种类主要包括 CpG7909(别称 CpG2006,CpG-2006)、ISS-1018、CpG684 等。其中 Dynavax 公司采用CpG 类佐剂(ISS-1018)的增效乙肝疫苗(Heplisav®)于2017 年9 月在美国批准上市。已有超百种含CpG 佐剂的治疗性疫苗及预防性疫苗进入临床试验阶段,30 多个品种使用了CpG7909。而在预防用疫苗制品中,佐剂使用情况主要包括CpG 单佐剂、CpG 佐剂加氢氧化铝或MF59 佐剂等,不同品种疫苗中CpG 临床用量为100 ~ 3 000 μg 不等,使用量集中在 250 ~ 1 000 μg / 剂,最大使用量为目前已批准上市的乙肝疫苗(Heplisav®),其临床用量为3 000 μg / 剂[9-15]。国内尚无已批准上市产品,已批准上临床的产品包括增效乙肝预防性疫苗等。
CpG 能显著增强抗乙肝、炭疽和流感等传染病的含铝佐剂类疫苗的免疫原性和加速免疫反应[4,16-17]。但CpG 佐剂会引起轻微至中度的副反应,包括注射位点疼痛、肿胀、硬结、红疹等局部反应以及流感样症状等系统症状,几天内会得到缓解,可能是由其免疫刺激特性导致的[18-20]。
3 预防用疫苗中CpG ODN 佐剂研究的整体考虑
CpG 作为一种新型佐剂,可参照欧盟及WHO 公布的《人用疫苗佐剂研究技术指南》、《含佐剂疫苗及疫苗佐剂技术指南》等[21-22]开展药学、临床前及临床研究。立题依据方面,疫苗开发过程中对CpG类别(序列)及用量的选择,可从CpG 免疫机制、临床应用及抗原特性、佐剂-抗原相互作用等方面进行综合考虑。免疫作用机制方面,可能需要考虑的因素包括疫苗可能发挥药效的作用机制(体液免疫、细胞免疫)及对应的佐剂类型、基序等,如TLR9 特异性识别的CpG 核心基序是由6 个碱基组成的,不同CpG 含有的核心基序存在差异,核心基序及其数量、间隔序列、整体长度和CpG 种属特异性的差异会导致免疫刺激活性的差别,从而影响制剂中佐剂用量。建议对设计的CpG 序列与人类基因组序列同源性进行比对研究,尤其是同源性对安全性的影响。临床应用方面,可参考已上市或临床试验阶段产品中使用情况,基于已获得的安全有效性数据进行评估选择。特定抗原方面,理论上不同的抗原表面电荷等性质可能会影响CpG-抗原间的相互作用,需结合免疫原性、吸附率、稳定性等方面进行综合考虑。作为疫苗佐剂CpG,应关注序列设计涉及的知识产权问题,应不构成专利侵权。
对于含CpG 佐剂的制剂处方、配制工艺、终产品质量特性、质量放行标准、稳定性等研究,可参考《预防用含铝佐剂疫苗技术指导原则》[23]及国外佐剂相关技术指南等要求开展。作为一种新型佐剂,通常需要考虑单独的佐剂非临床研究及制剂非临床研究,也需考虑完整的临床设计,包括剂量、免疫程序探索等。非临床研究阶段除常规非临床安全性、有效性研究外,建议考虑对CpG 在机体中的药代动力学等进行评估。在临床剂量探索及免疫原性研究中,建议针对抗原剂量、CpG 含量等进行体液和细胞免疫两方面的评估。
产品注册申报阶段,按照《生物制品注册分类及申报资料要求》[24]提供相关研究资料。作为佐剂应提供佐剂概述、佐剂完整的药学研究信息,包括原材料、工艺、质量属性、检测方法、稳定性等。非临床研究中如有药代、毒理学研究,按照ICH M4 基本框架在相应部分提交使用佐剂类型、添加佐剂必要性及佐剂/ 抗原配比合理性、佐剂机制等研究内容。
4 CpG ODN 佐剂的药学研究考虑
4.1 生产工艺开发 CpG 人工合成时为使其能够抵抗核酸酶,延长其在体内的作用时间,合成过程将对其进行全硫代化修饰,即骨架磷酸基团上的非桥氧原子替换为硫原子。CpG 生产工艺流程可能包括固相合成(包括耦合反应、硫化反应及乙酰化反应等)、氨解反应、纯化精制、脱盐浓缩等步骤及可能的冻干工艺。其中固相反应采用核酸合成仪按照设定的工艺参数自动完成。
CpG 生产应符合GMP 要求,生产过程中应研究确定关键工艺参数、中间品控制项目及质量标准等。对关键原材料如固相载体、核苷酸、合成用化学原料等来源和质量进行控制。应确定生产工艺及生产规模,关注生产规模与拟研制的疫苗生产规模的匹配性。根据研究拟定合理的产品放行质量标准,并在临床开发阶段不断予以完善,实际生产中应做好规范的制检记录。
4.2 质量特性研究 开发者应根据实际生产工艺及产品预期特性进行全面的研究。一般情况下,质量研究包括CpG 佐剂结构特性、理化性质、生物学活性及杂质谱等。
对于疫苗类制品,应对非目标成分进行控制。《中国药典》三部(2020 版)中已明确要求,生产过程中应尽可能减少使用对人体有毒、有害的材料,必须使用时,应验证后续工艺的去除效果[25]。除非验证结果提示工艺相关杂质的残留量远低于规定要求,且低于检测方法的检测限,通常应在成品检定或适宜的中间产物控制阶段设定该残留物的检定项。应通过工艺研究、工艺表征确定产品相关物质/杂质产生、去除的阶段及最终含量,并采用适宜的分析方法予以鉴定。应在成品检定或适宜的中间产物控制阶段进行制品相关物质/杂质的检测并设定可接受的限度要求。
根据CpG 生产工艺情况,杂质可能来自原料如原料单体 dATP、dTTP、dGTP 和 dCTP 以及保护剂和溶剂等,也可能来自反应副产物如非目标序列(如缩短的序列,延长或者多聚的序列以及硫代不完全序列)及生产或贮存过程中的降解产物等。产品相关杂质方面,可考虑采用液相色谱-质谱(liquid chromatography-mass spectrometry,LC-MS)法对杂质谱进行分析,全面收集信息,明确出现的各峰归属,以此为基础建立合理的质控标准。工艺相关杂质方面,杂质类别主要可能包括细菌内毒素、元素杂质及工艺步骤中添加的各种有机溶剂。应对热原、元素杂质、残留有机溶剂等进行研究分析。
另外,考虑到残留核酸酶和甲醛可能导致CpG的降解和交联,且抗原生产过程中工艺相关杂质、各种缓冲液可能引起CpG 相关杂质改变等,应开展充分的稳定性考察,重点关注是否存在降解、生物学活性下降等。
此外,还需对添加CpG 佐剂后的制剂开展充分的质量特性研究。包括CpG 佐剂与抗原及其他佐剂的相互作用情况(如是否吸附等),抗原及疫苗配方系统对CpG 的影响(如是否引起降解产生杂质,使生物学活性下降等),同时应研究CpG 佐剂与抗原的相互作用情况,在疫苗成品中的含量情况等。作为单链DNA,CpG 通常带负电,其与抗原的相互作用取决于抗原表面电荷、佐剂表面电荷(如配方中含其他佐剂)、缓冲液类型等因素。关于抗原相互作用研究,建议采用多种检测方法在多种条件下进行CpG与抗原相互作用的研究。检测方法包括还原/ 非还原电泳、毛细管电泳、层析及细胞检测法等,检测条件可考虑不同pH、离子强度、温度等因素。
4.3 质量标准 目前尚无CpG 质量标准被国内外药典收载。开发者应根据质量研究情况建立合理的原液、半成品及成品质量标准。
结合已有临床研究产品中对CpG 建立的质量标准情况,质控项目建议包括但不限于特性鉴别,如序列测定(测序或质谱法)、相对分子质量(质谱法)、硫代率(核磁共振法)、元素分析(C、H、N、P、S、Na 等主要元素);理化特性,如颜色及外观、pH、紫外最大吸收波长;纯度;杂质残留,如有机溶剂残留量、热原检查、微生物限度;生物学活性测定。此外,需关注疫苗成品中增加的相关检定项目,如CpG 含量测定、吸附率(如有吸附)及成品生物学活性等。
检定方法学方面,应参照国内外相关原则进行全面的方法学验证。以下对几种较为关键的检测方法进行讨论:
序列测定可采用核酸测序法及液相色谱-串联质谱法(liquid chromatography-mass spectrometry/mass spectrometry,LC-MS / MS),但由于 CpG 序列中核苷酸的重复序列较多,采用LC-MS / MS 法的测定结果并不能准确反映CpG 序列的正确性,更宜结合TA克隆核酸测序法进行综合的测序分析。硫代率影响CpG 的稳定性,是评价CpG 质量的重要指标,采用的检测方法应可区分硫代完全与硫代不完全的产品,如采用核磁磷谱法进行分析。元素杂质分析可参照ICH Q3D,采用ICPMS 法。纯度方法应充分验证,CpG 的潜在杂质包括氧化、硫代不完全等带来的杂质,不同色谱对不同杂质的分离效果不同,对于潜在杂质应进行多种原理的纯度控制,如广泛应用于核酸药物研究和纯度分析的强阴离子交换色谱法(strong anion exchange-high performance liquid chromatography,SAX-HPLC)[26]、反相高效液相色谱法等。
残留有机溶剂通常采用气相色谱法分析,依照《中国药典》四部(2020 版)等进行残留和毒性评估。对于《中国药典》四部(2020 版)已收载的溶剂,可参考相关溶剂分析方法及残留限度进行控制。对于《中国药典》四部(2020 版)、ICH 等指导文件中无限度规定和未收载的有机溶剂,可考虑通过查阅化学物质毒性数据库等资料,获得试剂的毒性研究数据,建立合适的质量标准进行控制。通常,有机溶剂残留质量标准限度与工艺能力应在同一数量级。
生物学活性测定方面,目前采用的方法包括基于免疫机理构建的HEK-Blue hTLR9 转基因细胞进行的报告基因法[27-28]及CpG 体外活化免疫细胞功能等方法,后一种方法是采用人树突状细胞系GEN细胞作为效应细胞,ELISA 法检测细胞分泌至上清中细胞因子情况,从而反映CpG 体外活化免疫细胞功能。在保证上述方法条件一致的情况下,特定细胞因子对应的CpG 半数有效浓度也可作为CpG 种类和序列筛选对比的临床前参考依据。对于体内免疫学活性测定,根据免疫学原理,Th2 细胞促进IgG1亚型抗体的分泌,Th1 细胞促进IgG2a 亚型抗体的分泌,因此IgG2a 亚型抗体水平在一定程度上可反映出机体免疫应答过程中Tn(naive T)细胞分化为Th1 细胞的趋势。如常用的铝佐剂主要增强Th2 细胞介导的适应性免疫应答,CpG 佐剂主要介导Th1 细胞介导的应答,通过检测免疫小鼠IgG2a 亚型抗体表达量情况,可反映CpG 佐剂体内活性情况。对CpG 的活性测定,重点关注建立的方法应尽可能反映作用机制及显著的量效关系,如纳入质控标准还需考虑检测方法的敏感度、精密度、耐用性等,最终综合评定,必要时建议开展检测方法与体内活性的相关性研究。
疫苗成品中CpG 含量测定为重要指标,应考虑采用专属性强的检测方法,如采用电感耦合等离子体原子发射光谱(inductively coupled plasma-atomic emission spectrometry,ICP-AES)或电感耦合等离子体质谱(inductively coupled plasma mass spectrometry,ICPMS)等方法,通过测定S、P 等的含量确定CpG 含量。如通过抗原相作用研究证明CpG 在疫苗系统中以吸附状态存在,需检测吸附率。
对于CpG 佐剂及含有CpG 佐剂的成品,其保存条件、运输方法及效期等需根据稳定性考察研究数据确定。稳定性考察方案总体上可参照《生物制品稳定性研究指导原则》,并根据产品特点结合保存、包装和运输等具体情况合理设计。建议考察生产工艺相关杂质、配制佐剂用溶液及疫苗成品缓冲液性质(如盐类别及浓度、pH、其他组分)对CpG 稳定性的影响,根据CpG 特性,选择的检测方法灵敏度应能足够监测CpG 降解情况,重点建议关注纯度、硫代率、含量、生物学活性、降解情况及吸附完全性等指标。
5 小 结
随着临床研究及应用数据的积累,CpG 类佐剂免疫机制得到越来越多的证实,成为疫苗研究的热点之一。不同于传统生物制品,CpG 佐剂多以化学合成方式生产,其生产和质控是合成、免疫、生物学等专业的综合体现,随着研究的广泛、深入开展,将更加关注对不同来源CpG 佐剂的安全性、有效性和质量可控性,研究数据的不断积累也将提升研究者与监管方对CpG 的认知。
