4种他达拉非杂质的合成
2021-12-21刘文涛李新志孔祥雨
张 彬,刘文涛,李新志,杨 利,孔祥雨
(山东省药学科学院 山东省化药重点实验室,山东 济南 250101)
他达拉非,化学名为:(6R,12aR)-6-(1, 3-苯并二恶茂-5-基)-2-甲基-2, 3, 6, 7, 12, 12a-六氢化吡嗪并[1’, 2’: 1, 6]-吡啶并[3, 4-b]吲哚-1, 4-二酮,分子式:C22H19N3O4,相对分子质量:389.41。起初由葛兰素史克公司[1]研发,随后被转让给美国艾科斯(ICOS)公司,后经ICOS公司与礼来公司联合开发,并于2003年11月经美国药品食品监督管理局(FDA)批准上市。他达拉非是 FDA 和中国国家食品药品监督管理总局(CFDA)批准的唯一长效5型磷酸二酯酶抑制剂(PDE5),其半衰期为17.5 h,比西地那非长[2],持续时间久,使用剂量小,心血管安全性好,不会增加心血管疾病的风险,具有良好的安全性和耐受性[3]。其适应症除勃起功能障碍(ED)外,分别于2009年5月和2011年10月经FDA批准用于治疗肺动脉高压[4-5]和良性前列腺增生[6]。此外,他达拉非还用于急性缺氧性肺血管收缩[7]和减少内脏脂肪蓄积[8]。2013年开始,他达拉非在销售额上超过西地那非,2015年实现23亿美元销售额,在市场份额和净销售额上全面超越其主要竞争对手,成为ED市场遥遥领先的全球领导者[9]。
他达拉非的合成需经与甲胺水溶液的环合反应[10],质量控制研究中发现4类与甲胺有关的杂质,包括杂质I((6R, 12aR)-6-(1, 3-苯并间二氧戊环-5-基)-2, 3, 6, 7, 12, 12a-六氢化吡嗪并[1’, 2’: 1, 6]-吡啶并[3, 4-b]吲哚-1, 4-二酮),杂质II((6R, 12aR)-6-(1, 3-苯并间二氧戊环-5-基)-2-乙基-2, 3, 6, 7, 12,12a-六氢化吡嗪并[1’, 2’: 1, 6]-吡啶并[3, 4-b]吲哚-1,4-二酮),杂质III((6R,12aR)-6-(1, 3-苯并间二氧戊环-5-基)-2-氨基-2, 3, 6, 7, 12, 12a-六氢化吡嗪并[1’, 2’: 1, 6]-吡啶并[3, 4-b]吲哚-1, 4-二酮),杂质IV((1R, 3R)-1-(1, 3-苯并二恶茂-5-基)-2-(2-(二甲氨基)乙酰基)-2, 3, 4, 9-四氢-1H-吡啶并[3, 4-b]吲哚-3-羧酸甲酯),这是由于甲胺的工业制备过程中不可避免地会含有其他氨类化合物,这些化合物不容易被检测出来,而且会与环合之前的他达拉非中间体发生反应,从而在他达拉非原料药中引入这4种杂质。这4种杂质的合成方法未见文献报道,故对这4种杂质进行了合成研究,以便作为杂质对照品,为他达拉非原料及其中间体的质量控制研究提供指导和帮助。4种杂质的结构见图1。

图1 杂质I、杂质II、杂质III、杂质IV结构
1 仪器与材料
1.1 仪器
Inova-400超导核磁共振波谱仪(美国Varian,内标TMS);Agilent 1260高效液相色谱仪(美国Agilent);LCQ型质谱仪(美国Finnigan);电子分析天平(瑞士Mettler Toledo)。
1.2 药品与试剂
N-氯乙酰代四氢-β-咔啉((1R, 3R)-1-(1, 3-苯并二恶茂-5-基)-2-(2-氯乙酰基)-2, 3, 4, 9-四氢-1氢-吡啶并[3, 4-b]吲哚-3-羧酸甲酯)(实验室自制[10]);30 %氨水(分析纯,莱阳市康德化工有限公司);70 %一乙胺溶液(分析纯,天津大茂化学试剂厂);80 %水合肼(分析纯,山东西亚试剂);40 %二甲胺水溶液(分析纯,天津大茂化学试剂厂);其余试剂均为分析纯。
2 方法与结果
2.1 杂质I的合成
合成路线见图2。
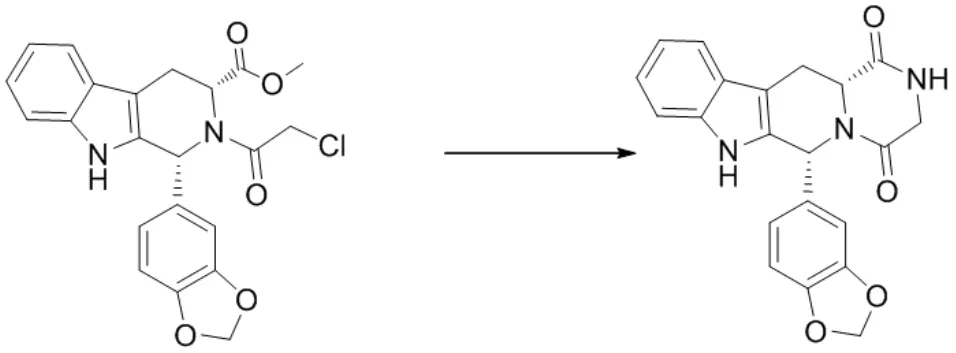
图2 杂质I的合成路线
取N-氯乙酰代四氢-β-咔啉(10 g,0.0235 mol),30 %氨水(35.5 g,0.305 mol),甲醇80 ml加入250 ml三口瓶中,搅拌加热至回流,回流反应30 h,HPLC监控至反应完全(N-氯乙酰代四氢-β-咔啉<1 %),过滤,得白色固体,加至甲醇-水(1:1)100 ml中室温搅拌2 h,过滤,滤饼用少量甲醇淋洗,60 ℃真空干燥,得白色固体杂质I 7.8 g,HPLC纯度99.3 %。ESI-MSm/z实测值(计算值):374.3(374.12)[M-H]-。1H NMR(400 MHz,DMSO-d6)δ:11.06(s,1H),7.91(m,1H),7.82(s,1H),7.35~7.12(m,3H),6.92~6.87(m,3H),6.18 (s,1H),5.95(d,2H),4.39(dd,1H),4.21~4.03(m,2H),3.75(dd,1H),3.23(dd,1H)。
2.2 杂质II的合成
合成路线见图3。
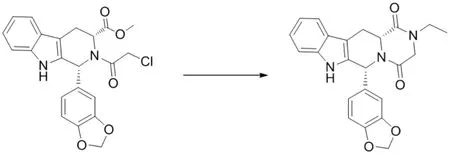
图3 杂质II的合成路线
将N-氯乙酰代四氢-β-咔啉(10 g,0.0235 mol),乙醇80 ml,70 %一乙胺溶液(3.98 g,0.062 mol)加入250 ml三口瓶中,搅拌加热至回流,回流反应10 h,HPLC监控反应至反应完全(N-氯乙酰代四氢-β-咔啉<1 %),降温至室温,过滤,得类白色固体,加至乙腈-水(2:1)100 ml中,室温搅拌2 h,过滤,少量乙腈淋洗滤饼,60 ℃真空干燥得白色固体杂质II 8.5 g,HPLC纯度99.5 %。ESI-MSm/z实测值(计算值):402.1(402.15)[M-H]-。1H NMR(400 MHz, DMSO-d6)δ:11.06(s,1H),7.82(s,1H),7.35~7.12(m,3H),6.92~6.87 (m,3H),6.18(s,1H),5.95(d,2H),4.41(dd,1H),4.21~4.03(m, 2H),3.92(m,2H),3.72(dd,1H),3.21(dd,1H),1.23(t,3H)。
2.3 杂质III的合成
合成路线见图4。
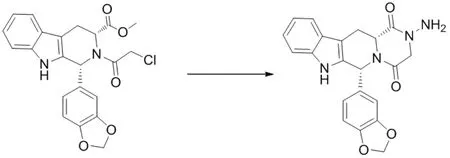
图4 杂质III的合成路线
将N-氯乙酰代四氢-β-咔啉(10 g,0.0235 mol),甲醇80 ml,80 %水合肼(5.135 g,0.082 mol)加入250 ml三口瓶中,搅拌加热至回流,回流反应8 h,HPLC监控反应至反应完全(N-氯乙酰代四氢-β-咔啉<1 %),降温至室温,过滤,得到类白色固体,加至甲醇-水(2:1)100 ml中,室温搅拌2 h,过滤,少量甲醇淋洗滤饼,60 ℃真空干燥得白色固体杂质Ⅲ 8.4 g,纯度99.2 %。ESI-MSm/z实测值(计算值):389.3(389.13)[M-H]-。1H NMR(400 MHz,DMSO-d6) δ:11.06(s,1H),7.82(s,1H),7.35~7.12(m,3H),6.92~6.87(m,3H), 6.18(s,1H),5.95(d,2H),4.39(dd,1H),4.30(s,2H),4.19~4.01(m,2H),3.75(dd,1H),3.23(dd,1H)。
2.4 杂质IV的合成
合成路线见图5。

图5 杂质IV的合成路线
将N-氯乙酰代四氢-β-咔啉(10 g,0.0235 mol),乙醇30 ml,纯化水10 ml,40 %二甲胺水溶液(23.9 g,0.212 mol)加入100 ml三口瓶中,搅拌加热至65 ℃,反应3 h,HPLC监控反应至反应完全(N-氯乙酰代四氢-β-咔啉<1 %),降温至室温,过滤,得白色固体,加至乙醇-水(1:1)100 ml中,室温搅拌2 h,过滤,少量乙醇淋洗滤饼,60 ℃真空干燥,得白色固体杂质IV 8.7 g,纯度99 %。ESI-MSm/z实测值(计算值):434.1(434.18)[M-H]-。1H NMR(400 MHz,DMSO-d6)δ:11.06(s,1H),7.81(s,1H),7.35~7.12(m,3H),6.92~6.87(m,3H),6.18(s,1H),5.95(d,2H),4.36(dd,1H),3.94(s,3H),3.75(s,2H),3.56(dd,1H),3.03(dd,1H),2.77(s,6H)。
3 结论
本文首次报道了他达拉非4种杂质的合成方法。以N-氯乙酰代四氢-β-咔啉为原料,分别与氨水,一乙胺,水合肼和二甲胺发生反应合成4种杂质,精制得到纯度都>99 %的目标产物。合成路线简单,收率高,原料易得。合成的4种杂质结构经MS和1H NMR确认,可作为他达拉非质量控制的杂质对照品。
