纤锌矿型β-CuGa1−xZnxO2的Zn2+取代Ga3+不等价掺杂对其结构及光催化性质的影响
2021-12-16姚明彩刘孝娟
姚明彩,杨 强,孟 健,刘孝娟
(1.稀土资源与利用国家重点实验室,中国科学院长春应用化学研究所,长春130022;2.中国科学技术大学应用化学与工程学院,合肥230026)
自Fujishima和Honda发现TiO2单晶可以光致水裂解产生H2和O2以来,光催化技术作为一种工艺简单、能耗低、操作条件易于控制、光催化材料易得、无二次污染的环保新技术在染料降解、重金属还原、水裂解、固氮和固碳等领域均得到了较为广泛的关注[1~3].光催化技术可直接利用太阳能驱动半导体材料表面的光氧化-还原反应,以期达到在室温下深度反应的效果.实际的光催化过程通常包含以下几个步骤:(1)光生载流子的生成;(2)光生载流子的分离和迁移;(3)到达表面生成活性物质降解有机污染物.其中,分离和迁移是决速步骤.这是因为电荷迁移过程通常需要几百ps,而缺陷、晶界和杂质离子都可能成为复合中心,以及热致载流子迁移的随机性,致使光生电子和光生空穴的复合往往只需几ps,远比电荷迁移快得多.因此,目前的半导体光催化剂还远不能满足实际应用的需要.而异质结制备、金属或金属氧化物装饰、表面缺陷工程等方面都能有效地促进表面电荷分离[4~6],但在体电荷分离方面进展缓慢.而近年来极化电场已被证明是一种改善光生载流子在半导体催化剂体相内和表面上分离的有效方法[7~9].极性广泛存在于非中心对称材料的内部,是由于空间反演破缺造成的正负电荷中心不重合从而产生自发极化,如电介质材料中的压电材料和铁电材料.极化电场作为直接的驱动力,可以促进光生电子(e−)和光生空穴(h+)的快速分离以及迁移,从而促进了各个位点的氧化还原反应快速发生.另外,极化电场也可以加速半导体催化剂的表面电荷与H2O和O2之间的能量转换,促进活性物质(如O2‧-,OH‧等)的生成[10,11].为了满足成本和 环境友好的要求,当制备器件时,低毒性和储量丰富的材料需要被优先考虑.基于以上考虑,氧化物可以作为主要备选材料,它们具有以下优势:在大气环境中通常很稳定,储量丰富,较硫化物和硒化物更安全,制备过程也更为简单环保.因此,越来越多的研究人员致力于寻找新型的氧化物半导体材料.
纤锌矿衍生的CuGaO2(β-CuGaO2),作为一种带隙约为1.5 eV新型的氧化物半导体已经被报道合成[12].理论计算认为β-CuGaO2也是一种具有较大自发极化的铁电体,自发极化值为83.80 μC/cm2[13],而β-CuGaO2作为铁电体已被Yao等[14]的实验证实.作为一种新颖的本征窄带隙铁电半导体,β-CuGaO2在光电器件制备中较传统的氧化物半导体有以下优势:(1)光能的利用率高.光能的损失通常有两部分组成:太阳光的吸收效率低,这是由于氧化物半导体通常是宽带隙半导体,大多只能吸收紫外光或部分紫外光,而太阳光谱的能量分布主要由可见光(约50%)和红外光(约43%)组成,紫外光只占很小的一部分(约7%);半导体中大部分的光生电子-空穴对都会复合,并以热能损失掉.这很大程度上是由光生电子空穴迁移的随机性造成的.通常构建异质结的方法可改善这一问题,但内建电场只分布在异质结附近,在材料体相内的迁移问题依然无法解决.而铁电体的自发极化电场是一种体效应,均匀地分布在整个材料内部,很大程度上可以解决光生电子和空穴随机迁移造成的复合问题.(2)光电器件效率高.以太阳能电池为例,器件功率基本由开路电压和短路电流决定.对半导体材料而言,开路电压最大值由材料的带隙值决定,短路电流的最大值受材料的带隙值影响.较大的开路电压需要宽带隙,较大的短路电流需要窄带隙,两者之间看似存在不可调和的矛盾.而铁电光伏的光生电压不受带隙影响,仅由材料的极化程度决定,甚至可以产生比带隙值高几个数量级的开路电压,因此可同时拥有较大的开路电压和短路电流的潜力,从而可能获得高功率的单结太阳能电池.β-CuGaO2是一种p型的亚稳相黑色粉末状功能陶瓷材料,无法适应很多潜在的应用环境.基于晶体结构和晶格失配度的考虑,本文在Ga-位引入不等价的Zn2+离子,合成了Zn掺杂的β-CuGa1−xZnxO2(x=0.03,0.05,0.07,0.10)体系,探究了Zn2+对β-CuGaO2的晶体结构、热力学稳定性、光学吸收以及光催化性质的影响.
1 实验部分
1.1 试剂与仪器
Ga2O3(纯度99.999%)购自麦克林试剂公司;Na2CO3(纯度99.999%)和CuCl(纯度99%)购自阿拉丁试剂公司;ZnO(纯度99.99%)购自国药基团化学试剂有限公司;柠檬酸(纯度99.5%)和聚乙烯醇(分子量20000)购自西陇化学试剂有限公司;甲基橙(分析纯)购自北联精细化学品开发有限公司.
Bruker D8型X射线衍射仪(XRD)和D8 Advance型原位高温X射线衍射仪(HT-XRD)(德国Bruker AXS公司);Tecnai G2F-20型热场发射透射电子显微镜(TEM,美国FEI公司);UV-3600型紫外-可见分光光度计(UV-Vis,日本岛津公司);STA 449 F3型同步热分析仪(TG-DSC,德国耐驰仪器公司);ESCALAB 250型X射线光电子能谱仪(XPS,美国Thermo公司);Sun 2000型太阳光模拟器(AM1.5,美国ABET Technologies公司);Fluorolog 3模块式荧光光谱仪(PL,法国Horiba Jobin Yvon公司).
1.2 样品的制备
主要采用金属离子交换法[12,15]制备该系列功能陶瓷.实验原料主要为Ga2O3,Na2CO3,ZnO和CuCl.按照化学式NaGa1−xO2(x为Zn的掺杂量,x=0.03,0.05,0.07,0.10)称量Ga2O3和Na2CO3,置于锥形瓶中并加入硝酸溶解.待全溶后,按照与金属离子1∶1~1∶1.2的比例加入柠檬酸和聚乙烯醇,转移至水浴锅上,水浴蒸发后制成干凝胶.将所得干凝胶研磨成粉,称量,计算所得干凝胶的摩尔数,加入所需的ZnO,研磨5 h,压制成片,在1500℃下煅烧4 h形成NaGa1−xZnxO2前驱体粉末.称量前驱体的质量,按照Na离子和Cu离子摩尔比1∶1添加适量的CuCl粉末,研磨24 h,并将研磨好的样品转移至管式炉内,在Ar气氛中于150℃下煅烧240 h.然后,用丙酮、二氯甲烷、乙酸乙酯、乙醚和去离子水等以体积比1∶2∶4∶1∶2配成的混合溶剂洗涤样品,去除NaCl和其它可溶性杂质.最后,使用真空干燥箱干燥样品,取出研磨,待用.
1.3 第一性原理计算
计算均基于密度泛函理论(DFT),采用广义梯度近似GGA+U的方法,由VASP软件包执行完成[16~18],其中,交换关联采用PBE函数[19].选取O,Cu,Zn和Ga的价电子结构分别是2s22p4(6e-),3d104s1(11e-),3d104s2(12e-)和3d104s24p1(13e-).为了匹配实验观察得到的晶体结构,Cu的3d电子,U值以0.5 eV为间隔从0 eV逐渐调至12 eV.Zn的3d电子也采用与Cu相似的方法.采用Gamma型5×4×5的k点对布里渊区进行积分,进行结构弛豫;接着在收敛计算后,采用Gamma型10×9×10的k点对布里渊区进行积分,进行态密度(DOS)、功函数和光吸收计算.平面波的截断能在结构弛豫时是520 eV,在DOS、功函数和光吸收计算时取400 eV.几何结构的弛豫标准是每个原子上的力小于0.1 eV/nm.结果显示,计算所得的初始晶格参数(a=0.55133 nm,b=0.66599 nm,c=0.53702 nm)与实验结果相符[20].实际计算模型中,DOS、能带结构和光吸收选用2×1×2的超胞,Zn的浓度为1/14(约为7%),Zn原子位置为随机选取;功函数选用2×1×2超胞,界面取向为(001),真空层1.5 nm,Zn原子位置选取真空层以下第四层的Ga原子(尽量减小掺杂原子对界面的影响);运用Berry Phase(BP)方法计算铁电极化,模型选用2×2×1的超胞,Zn的浓度分别为1/16(0.0625)和1/8(0.125).
1.4 催化降解实验
将25 mg催化剂分散于50 mL 100 mg/L甲基橙(MO)溶液中,以转速为200 r/min进行搅拌,使之均匀分散.无光照的降解在暗室中进行.光照下的降解置于光强为100 mW/cm2的模拟太阳光(AM1.5)下照射并保持搅拌,每间隔一段时间从降解池中取出1 mL液体,静置离心后取上层清液,并测量该上层清液的紫外-可见吸收光谱.根据MO分子在462 nm处的最大吸收值计算其降解率(η,%):

式中:c0(mg/L)为催化剂分散均匀后MO的浓度;c(mg/L)为MO在反应中t(min)时刻的浓度.
2 结果与讨论
2.1 晶体结构的表征
图1 为β-CuGa1−xZnxO2陶瓷粉末的XRD谱图.对于纯的β-CuGaO2陶瓷粉末,晶型为正交相结构,各个衍射峰的位置、强度与文献[21]报道一致,且未观察到杂质峰,表明已得到正交纤锌矿型纳米颗粒.由于Zn2+的半径(0.060 nm)与Ga3+的半径(0.047 nm)存在差别,通过XRD局部放大图可见,Zn2+的掺杂量≤0.05时,β-CuGaO2的(121)晶面衍射峰的位置随Zn2+掺杂量的增加向小角度方向偏移,说明掺杂的Zn2+成功替换了β-CuGaO2晶格中的Ga3+.值得注意的是,当Zn2+的掺杂量达到0.07时,β-CuGaO2的(121)晶面衍射峰的位置从36.08°偏移到36.05°后,基本不变,说明过多的Zn2+不再替换Ga3+,这和Zn2+与Ga3+不等价造成的电荷失衡有关.同时,在β-CuGa0.93Zn0.07O2样品中观察到了第二相(100)和(101)的衍射峰,符合六方的ZnO(PDF#75-0576)相的衍射峰特征,表明Zn2+浓度过高时形成第二相,即六方的ZnO相.
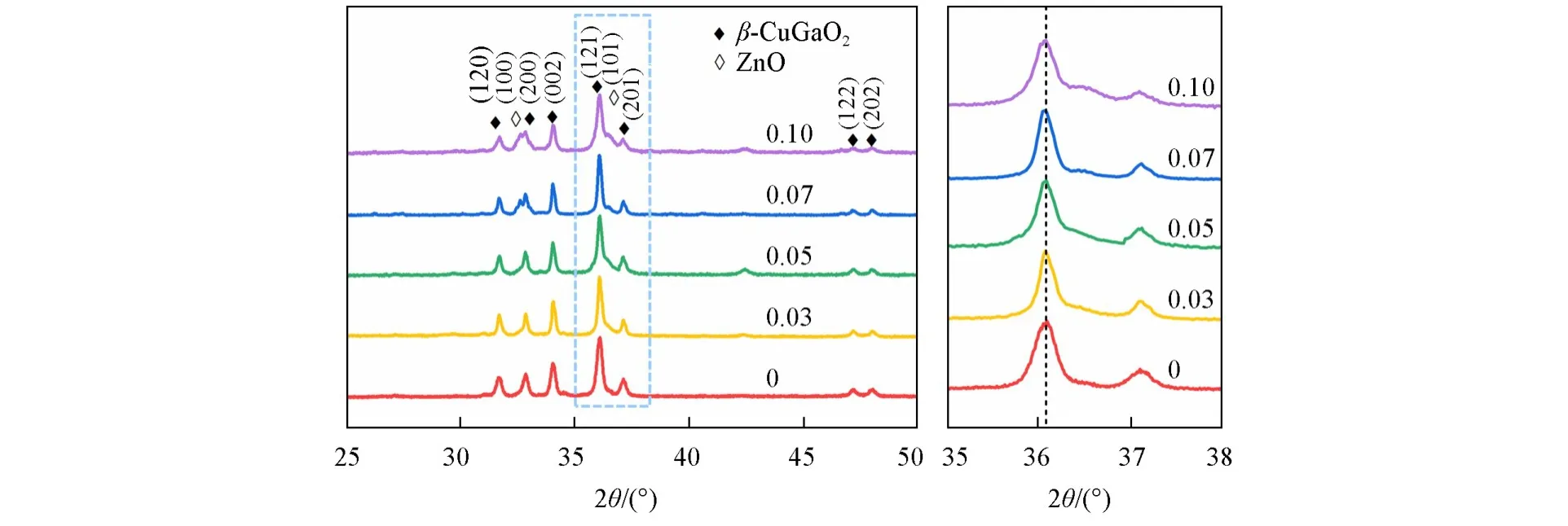
Fig.1 XRD patterns of β⁃CuGa1-xZnxO2(x=0.03,0.05,0.07,0.10)
由高角度环形暗场-透射电子显微镜(HAADF-TEM)照片[图2(A)和图3(A)]可知,本体相的β-CuGaO2呈现一定的规则形状,随着Zn2+离子的加入,规则的形状逐渐消失,而β-CuGa0.95Zn0.05O2大多形成不规则的块状物体,但材料中的各种元素始终均匀分布.由X射线能谱分析(EDS)的元素面分布[图2(B)~(D)和图3(B)~(D)]可见,Zn2+离子的引入降低了β-CuGaO2的结晶度,使其有从晶态向非晶态发展的趋势,与图1中掺杂后衍射峰展宽的趋势一致.由图2(E)可见,衍射晶面(121−)和(21−1−)的晶面间距分别为0.2003和0.2032 nm,两个衍射晶面的夹角为70.88°;由图3(F)可见,衍射晶面(111)和(020)的晶面间距分别为0.3047和0.3105 nm,两个衍射晶面的夹角为59.60°.图2(E)和图3(E)插图的选区电子衍射(SAED)表明,两者的点阵同属正交晶系的Pna21空间群,拟合的起始晶胞参数均为a=0.5513 nm,b=0.6560 nm,c=0.5270 nm,与文献[22,23]相符.

Fig.2 HAADF⁃TEM image(A),EDS elemental mapping of Cu(B),Ga(C),O(D),SAED pattern(E)of β⁃CuGaO2nanoparticles

Fig.3 HAADF⁃TEM image(A),EDS elemental mapping of Ga(B),Cu(C),Zn(D),HRTEM image(E)and HRTEM image after Fourier transform(F)of β⁃CuGa1-xZnxO2 nanoparticles
2.2 热稳定性分析

Fig.4 TG⁃DSC curves of β⁃CuGa1-xZnxO2 in Ar
图4 为Ar气中β-CuGa1−xZnxO2的TG-DSC测试结果.可见,本体相β-CuGaO2的相转变点在510℃[图4(A)].当x=0.05时没有出现第二相,相转变温度降为490℃[图4(B)],这很可能是由于用Zn2+替代Ga3+的不等价掺杂,引入了较多的氧空位,从而增加了晶格结构的不稳定性.当x=0.10时出现第二相,相转变温度又升为498℃[图4(C)],表明第二相的出现有利于纤锌矿结构的稳定.因为β-CuGaO2是ZnO的三元衍生物,两者晶体结构相似,晶格失配度较小,所以,CuGaO2容易从β相转变到α相[图5(A)和(B)].在Zn2+的过量掺杂形成ZnO的第二相时,局域聚集的ZnO通过掺杂量的增加,逐渐降低晶体结构整体的畸变程度,形成具有较高稳定性的三维空间晶体网,降低声子振动的平均振幅,在一定程度上可提高β-CuGaO2的热稳定性[图5(C)].因此,可通过制备固溶体提高β-CuGaO2-ZnO复合材料的热稳定性.
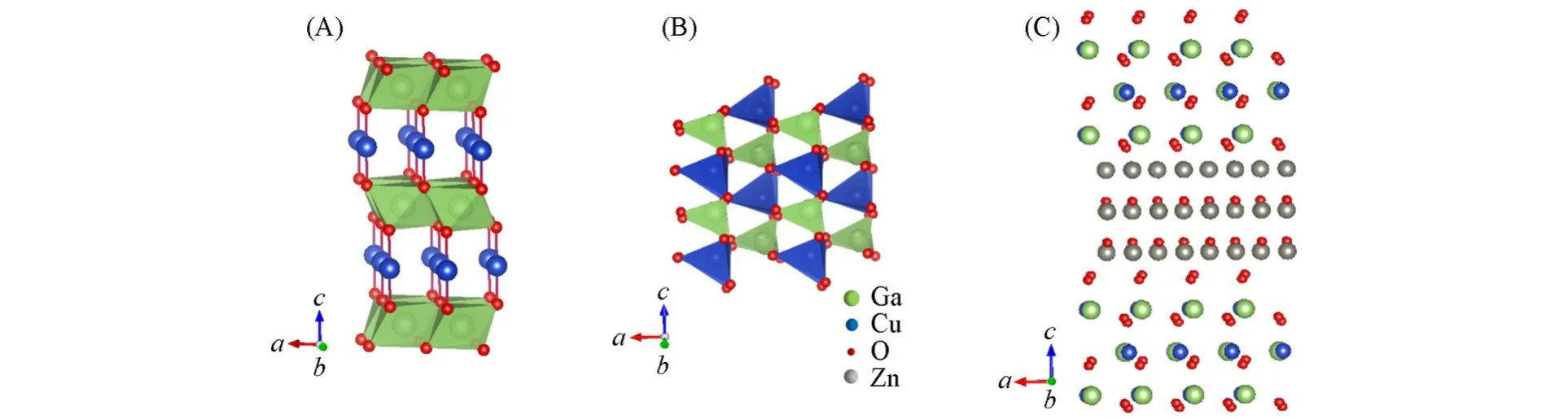
Fig.5 Crystal structures of α⁃CuGaO2(A)and β⁃CuGaO2(B),the sketch of the solid solution phase structure of β⁃CuGaO2 and ZnO(C)
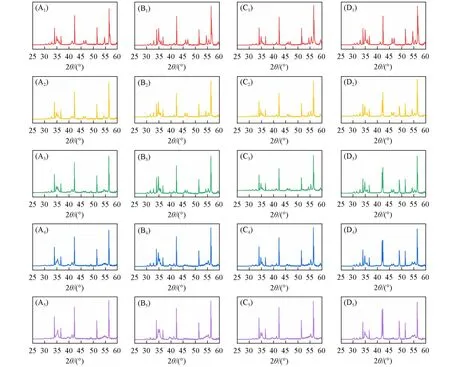
Fig.6 In⁃situ HT⁃XRD patterns of β⁃CuGa1-xZnxO2
图6 为在真空下β-CuGa1−xZnxO2的HT-XRD谱图.由图6(A)和(B)可知,当掺杂量≤0.05时,在260℃时,在2θ=36°附近已有杂质相的衍射峰.在280℃时,β-CuGa0.97Zn0.03O2的HT-XRD谱图中只有α-CuGaO2和ZnO的衍射峰,基本完成了由β相到α相的转变.此时β-CuGa0.95Zn0.05O2的HT-XRD谱图中β-CuGaO2依旧为主要衍射峰,这表明低浓度的不等价掺杂不利于热稳定性的提高.由图6(C)和(D)可见,当掺杂量x>0.05时,在310℃附近才出现杂质相的衍射峰,β-CuGa0.90Zn0.10O2在2θ=36°附近的杂质峰的峰高比β-CuGa0.93Zn0.07O2的更小,表明第二相的出现可能有利于降低声子的平均振幅,从而提高材料的热力学相变的温度.
图7 为β-CuGa1−xZnxO2的XPS谱图.研究发现,Cu2p3/2可劈裂成p1和p2两个峰,结合能933.02 eV处的p1峰应归属于Cu+,结合能934.62 eV处的p2峰应归属于Cu2+,其中Cu2+的卫星峰s1和s2的结合能分别为941.20和944.02 eV[24].考虑到制备方法及原料的稳定性,β-CuGaO2中不可避免会存在Cu2+,但含量通常较低[见图7(A)].Zn2+掺杂在Ga-位后,Cu2+峰面积变大,含量增加[见图7(B)].这可能是由于正电荷数量减少,氧空位的数量不足以维持材料内的电荷平衡,从而引起的Cu+升高价态成为Cu2+,以维持体相材料内的电荷平衡.因此在出现第二相之前,随着Zn含量的增加,晶格内电荷不平衡的程度在加剧,以致降低了材料的相变温度.

Fig.7 XPS spectra of β⁃CuGa1-xZnxO2
2.3 光学性质分析
图8 给出了β-CuGa1−xZnxO2的实验和计算光学吸收谱图.由图8(A)可知,随着Zn2+含量的增加,β-CuGa1−xZnxO2中第二段的光吸收越来越明显,比例越来越大.出现第二相之前,第二段吸收带边随Zn2+掺杂量的增加逐渐蓝移,直至分相后第二段的吸收带边逐渐趋于稳定值,但区别于纯的ZnO相.ZnO的吸收带边在380 nm附近,具有明显的紫外吸收特征.而出现的第二相具有典型的可见光吸收特征,这表明该第二相可能是以ZnO为溶剂,β-CuGaO2为溶质随机排列形成的纤锌矿型固溶相,而不是有序排列形成的掺杂晶体,并且该固溶相吸收带边受氧空位影响较大[25].使用第一性原理计算对Zn2+掺杂β-CuGaO2的吸收光谱进行了系统研究.由图8(B)可见,本征态吸收曲线陡峭,呈现直接带隙的特征;掺杂态吸收曲线多平台且较缓,有部分间接带隙的特征.表明光子的跃迁效率有可能降低.
图9 为根据图8(B)的模拟所做的计算,具体模型如图9(I)所示.根据功函数[图9(A)和(E)]和能级[图9(C)和(G)]可知,Zn2+替代Ga3+掺杂以后,价带顶(VBM)和导带底(CBM)均有所下移,但是VBM下移的程度更多,这与ZnO的能级较深有关[26].根据Mulikken电负性,结合半导体导带电势(ECB)和价带电势(EVB)的经验公式可得[27,28]:

Fig.8 UV⁃Vis diffuse reflection absorption curves of β⁃CuGa1-xZnxO2 of experimental(A)and calculated(B)

式中:χTol为绝对电负性;Ee是氢标下自由电子的能量(约4.5 eV);Eg为半导体的带隙.对于化合物AaBbCc,其绝对电负性可根据以下方法进行计算[29]:

式中:a,b,c分别为元素A,B,C的原子个数;χaA为A元素电负性的a次方,其中χA由该元素的电子亲和能以及电子解离能的平均值决定.查表计算可得[30],χGa是3.21 eV,而χZn是4.41 eV,因此Zn元素在Ga位的掺杂可使能带整体向更正的方向移动(向下移动),与图9(D)和(H)的计算结果一致.从DOS图可知,Zn2+引入以后,费米能级附近的价带和导带的电子密度还主要由Cu和O贡献,Zn的电子密度在价带的深能级中有所显现,但是能带图所展示的带隙还是稍有增加[图9(B)和(F)].值得注意的是,Zn2+掺杂以后,与之相近的Cu+被诱导出高于1的价态,出现自旋劈裂.但是这个自旋劈裂出现在价带的深能级处,远离费米能级,可认为不影响光电反应.根据经验公式可知,此时光学吸收带边从本征态的867 nm蓝移到掺杂态的821 nm.在半导体光催化中,光生电子具有强还原性,光生空穴具有强氧化性,VBM下移有利于增强空穴的氧化能力.因此,与β-CuGaO2相比,β-CuGa1-xZnxO2应该有更好的光氧化能力,理论上更有利于氧化降解反应的发生.

Fig.9 Property calculation of workfunction(A),DOS(B),energy band structure(up)(C)and energy level position(D)of β⁃CuGaO2,the property calculation of workfunction(E),DOS(F),energy band struc⁃ture(up)(G),energy level position(H)and calculated crystal model(I)of β⁃CuGa1-xZnxO2
2.4 降解反应及原理
运用降解偶氮类染料MO来探究Zn2+的掺杂对β-CuGa1−xZnxO2在实际应用中的影响.图10(A)和(B)分别为β-CuGa1−xZnxO2在无和有光照条件下降解有机物随时间变化的降解效率曲线.可见,Zn2+掺杂以后,无论是有光条件还是无光条件下,降解效率均较β-CuGaO2有所降低,表明Zn2+掺杂不利于降解反应,这与光学性质分析的结论相反.之所以出现“性质分析预测和实验结果相反”的情况,是因为两种条件下的反应机理不同.无光照时为极化降解,以极化电场为主要降解源动力,包含铁电极化和压电极化,如图11(A)所示.压电极化,是铁电极化在外场作用下的一种响应.压电催化可以由不同形式的机械能引起,如搅拌、声波、潮汐、风和大气压等.机械力(如超声)持续作用于液体,如水分子.这些水分子在压力(如声波)的波谷处被挤压,在波峰处被撕裂,从而产生大量的空化气泡[31].这些空化气泡到达临界尺寸(通常为几十微米)坍塌时,会对分散的压电材料产生较大的压力,甚至可高达108~109Pa[32,33].在这些空气泡经历形成、生长、破裂的过程中,压电诱导的电位还会周期性地出现,Yao等[14]在探究β-CuGaO2的降解机理时也印证了该观点.对于高绝缘的电介质而言,传导电流是可忽略的.因此,对于理想电介质,以下模型只考虑位移电流.位移电流密度(JD)定义为
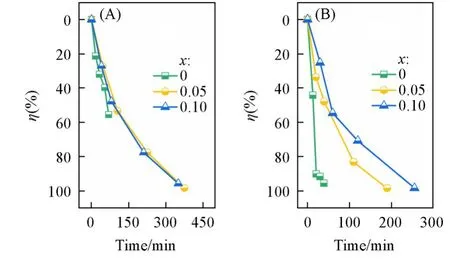
Fig.10 Degradation efficiency(η)of β⁃CuGa1-xZnxO2 under dark(A)and light(B)

Fig.11 Mechanism diagram of degradation reaction under dark(A)and light(B)
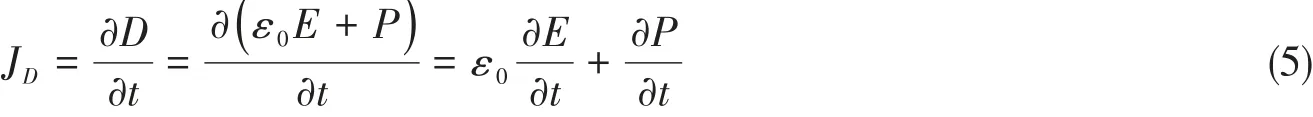
式中:ε0为真空介电常数;P为电介质材料中的电极化;E(N/C)为电场强度;t(s)为时间.由式(5)可见,位移电流密度是由P和E随时间的瞬态变化决定的.另外,由于没有外加电场,式(5)右侧第一项中的E来自于表面极化电荷产生的退极化场,第二项(∂P/∂t)称为极化电流,其是由介质材料中正电荷和负电荷的分离而产生的.显然,在β-CuGaO2纳米粒子内部,JD主要由(∂P/∂t)组成.但是,对于H2O分子包围的β-CuGaO2纳米颗粒,JD主要来自H2O诱导的退极化场,即


Fig.12 Band structures of spontaneous polarization state(A)and enhanced polarization state in external field(B)under ferroelectric domains
如此产生的电流会在颗粒两端产生高压电场[图12(A)],可以通过吸附溶液中的带电粒子增强该电场强度,增加β-CuGaO2的极化程度,进而使初始极化态的能带发生更大的倾斜,如图12(B)所示.Yao等[14]通过外加电场调节铁电畴的取向,使暗态下的降解效率成倍增加.这是因为压电催化降解过程遵循一级动力学反应,降解过程的动力学可以表示为ln(c0/c)随时间(t)的线性关系.无光照时,β-CuGa0.95Zn0.05O2和β-CuGa0.90Zn0.10O2的降解速率均小于β-CuGaO2,这表明Zn2+的掺杂可以降低材料的自发极化程度.Berry phase(BP)相计算结果证明了上述观点,β-CuGaO2的自发极化是98.22 μC/cm2,如图13所示,掺杂以后自发极化先降至81.99 μC/cm2(β-CuGa0.9375Zn0.0625O2),掺杂浓度再增加又降至71.22 μC/cm2(β-CuGa0.875Zn0.125O2),表明随Zn掺杂量的增加自发极化逐渐减小,两者呈现负相关关系.这主要是因为Zn2+的半径(0.060 nm)比Ga3+的半径(0.047 nm)大,缩小了与Cu+的半径(0.060 nm)的差距,减小了晶体内的偶极矩.而一旦光照,自由的光生电子-空穴对会在极化电场的作用下定向迁移,如图11(B)所示.因此有光照时,极化降解和光降解相辅相成,极化电场为主要驱动力,协助光生载流子的分离及迁移.在图11(B)中,随着Zn2+掺杂量的增加,β-CuGa0.90Zn0.10O2的降解速率比β-CuGa0.95Zn0.05O2更慢.这主要是因为虽然掺杂浓度对极化电场的影响明显,同时对光吸收影响较大.如图8(A)所示,随着Zn2+浓度的增加,本征态的光吸收带边逐渐蓝移,且比例减小,而掺杂态的光吸收带边也逐渐蓝移,且比例逐渐增大.意味着随着Zn2+浓度的提高,β-CuGa1−xZnxO2的吸光效率在下降,同等条件下产生的光生载流子的量在逐渐减少.此外,Zn2+含量越高,材料中的负荷中心数可能越多.图14给出了β-CuGa1−xZnxO2的光致发光光谱.可见,随着Zn2+掺杂量的增加,峰强度逐渐增强,表明β-CuGa1−xZnxO2载流子的复合速率在加快.这可能是由于:一方面,ZnO的激子结合能较高[34,35];另一方面,随着Zn2+掺杂量的增加,氧空位的量也逐渐增加,可导致材料内部的复合位点增加,致使复合速率增加.这些不利于器件对太阳光能的利用,会使光能以热能的形式损失掉,造成光能利用率的降低.
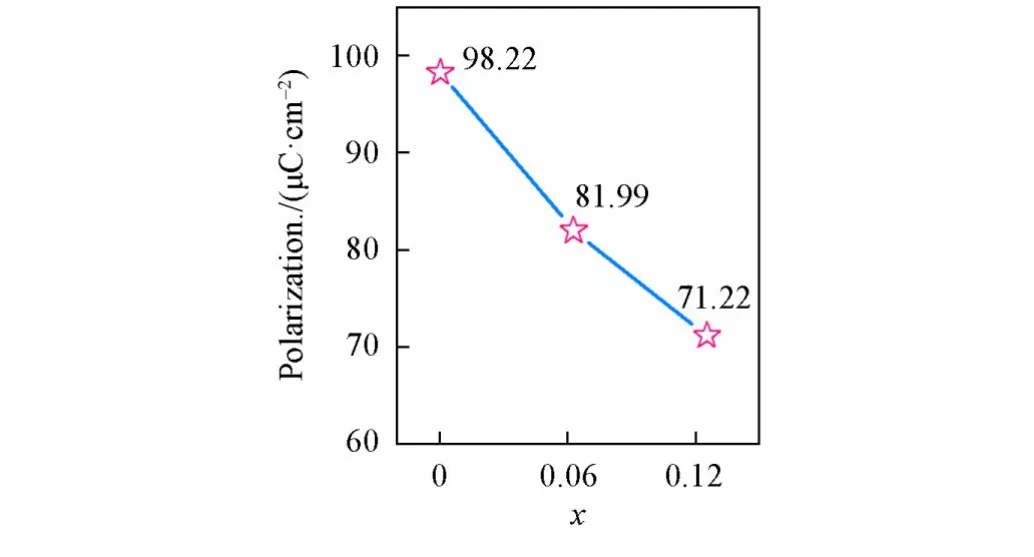
Fig.13 Macroscopical polarization data of Berry phase(BP)calculation models of β⁃CuGaO2,β⁃CuGa0.9375Zn0.0625O2 and β⁃CuGa0.875Zn0.125O2

Fig.14 PL spectra of β⁃CuGa1-xZnxO2
3 结 论
采用简单的金属离子交换法合成了β-CuGa1−xZnxO2系列材料,研究了Zn的不等价掺杂对β-CuGa1−xZnxO2材料结构及性质的影响规律,主要包括晶体结构、热力学稳定性和光学性质,并通过MO的降解反应对以上影响进行了评估.结果表明,Zn2+在Ga-位的低浓度不等价掺杂不利于该功能陶瓷的相变温度提高,较高浓度掺杂使相变温度稍有回升,这对提高该功能陶瓷的相变温度具有方向性的指导意义.由于Zn2+在Ga-位的不等价掺杂对自发极化和光吸收均有较为明显的不利影响,且Zn2+的深能级不足以弥补该不利影响,因此β-CuGa1−xZnxO2不利于光电反应的发生.
