一测多评法同时测定补中益气丸中6 种成分的含量
2021-12-15张永昕李莎恩蔡琴琴段付军
张永昕,李莎恩,蔡琴琴,张 莹,段付军
(中国人民解放军南部战区空军医院,广东 广州 510602)
补中益气丸由黄芪、党参、甘草、白术、当归、陈皮、升麻、柴胡组方[1],具有补气健脾、升陷利湿功效,对免疫系统、消化系统及代谢相关疾病均有积极的治疗作用[2-4]。补中益气丸的质量控制研究多为某一有效成分的含量测定,虽有多组分的测定研究,但该制剂成分复杂,操作过程烦琐,所需对照品较多,成本较高,不易开展[5]。一测多评(QAMS)法是一种用于多成分质量控制的分析方法,适用于评价中药多组分的质量[6-8]。本课题组在前期的薄层色谱和指纹图谱研究的基础上[9-10],建立了同时测定补中益气丸中黄芪有效成分毛蕊异黄酮葡萄糖苷[11],甘草有效成分甘草苷、甘草酸铵[12],陈皮有效成分橙皮苷[13],升麻、当归有效成分阿魏酸、异阿魏酸[14]6 种成分含量的QAMS 法,旨在为补中益气丸的质量控制提供科学、有效的新方法。现报道如下。
1 仪器与试药
1.1 仪器
P680 型高效液相色谱仪(美国 Dionex 公司);LPG-3400SDN 型高效液相色谱仪(美国Thermo Fisher Scientific 公司);CPA225D 型电子分析天平(北京赛多利斯天平有限公司,精度为十万分之一);KQ-400KDE 型超声清洗器(昆山市超声仪器有限公司,功率为400 W,频率为 40 kHz)。
1.2 试药
橙皮苷对照品(批号为C-006-150318-4,纯度为 99.0%),甘草苷对照品(批号为 G-009-141110,纯度为 98.7% ),阿魏酸对照品(批号为 A -002-140727,纯度为99.6%),异阿魏酸对照品(批号为Y-044-140801,纯度为99.0%),毛蕊异黄酮葡萄糖苷对照品(批号为 M -020-150130-1,纯度为 98.5%),甘草酸铵对照品(批号为G-003-150314,纯度为98.3%),均购自成都瑞芬思生物科技有限公司;黄芪、党参、甘草、白术、当归、陈皮、升麻、柴胡(广州至信中药饮片有限公司,批号均为20160711);补中益气丸(北京同仁堂制药厂,批号分别为 13061479,14080835,14082972,15071479,15080026,15080047,15081358,15082109,15082417,15082679,15111047,16080747);乙腈、甲醇均为色谱纯,其他试剂均为分析纯,超纯水经U 系列纯水机制备。
2 方法与结果
2.1 色谱条件与系统适用性试验
色谱柱:Diamosil C18柱(250 mm × 4.6 mm,5 μm);流动相:乙腈(A)-0.1%磷酸水溶液(B),梯度洗脱程序见表 1;流速:0.8 mL /min;柱温:30 ℃ ;检测波长:270 nm;进样量:10 μL。在此色谱条件下,橙皮苷、毛蕊异黄酮葡萄糖苷、异阿魏酸、甘草苷、阿魏酸、甘草酸铵与其他成分均可达到基线分离,分离度均大于1.5,理论板数按目标成分色谱峰计均大于4 000。色谱图见图1。
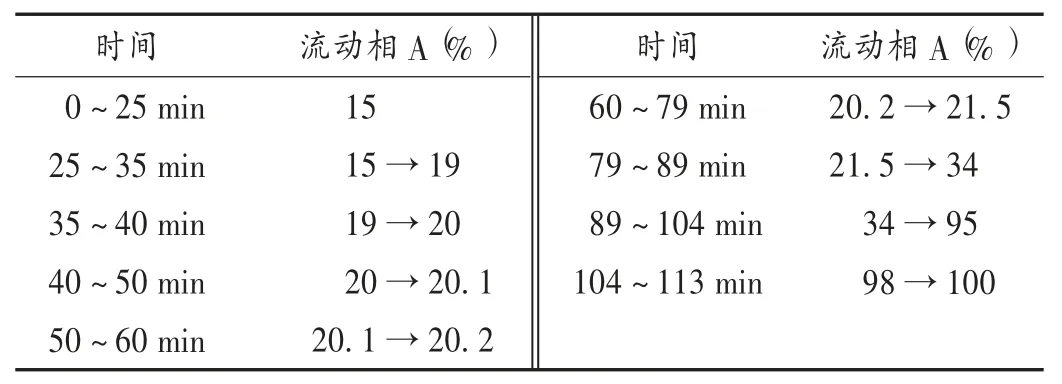
表1 流动相梯度洗脱程序Tab.1 The elution program of the mobile phase
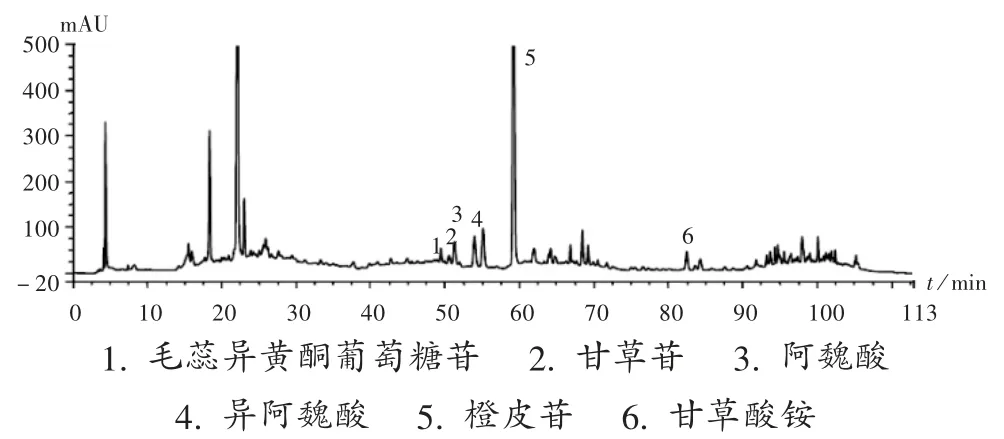
图1 系统适用性试验高效液相色谱图1.Calycosin - 7 - O - β - D - glucoside 2. Liquiritin 3.Ferulic acid 4.Isoferulic acid 5.Hesperidin 6.Ammonium glycyrrhizinateFig.1 HPLC chromatograms
2.2 溶液制备
取橙皮苷、毛蕊异黄酮葡萄糖苷、异阿魏酸、甘草苷、阿魏酸、甘草酸铵对照品各适量,精密称定,加甲醇适量使溶解,制成质量浓度分别为毛蕊异黄酮葡萄糖苷0.728 6 mg/mL、甘草苷 1.040 0 mg/mL、甘草酸铵1.000 0 mg/mL、阿魏酸 1.000 0 mg/mL、异阿魏酸1.040 0 mg /mL、橙皮苷 0.400 0 mg /mL 的单成分标准液。取各标准溶液适量,加甲醇制成毛蕊异黄酮葡萄糖苷、甘草苷、甘草酸铵、阿魏酸、异阿魏酸、橙皮苷质量浓度 分 别 为 3.643,15.600,57.000,1.500,15.600,120.000 μg /mL 的混合对照品溶液。取样品适量,研细,过80 目筛,取细粉2.0 g,精密称定,至具塞锥形瓶中,精密加入100%甲醇100 mL,称定质量,60 ℃超声提取30 min,共2 次,静置至室温,再称定质量,用100%甲醇补足减失的质量,摇匀,过滤,回收部分甲醇,将药液转移至100 mL 容量瓶中,定容,摇匀,即得供试品溶液。按处方比例,分别制备缺陈皮、甘草、黄芪、升麻和当归的阴性样品,依法制备缺陈皮、甘草、黄芪、升麻和当归的阴性对照品溶液。
2.3 方法学考察
专属性试验:取 2.2 项下溶液,按 2.1 项下色谱条件进样测定,记录色谱图,见图2。结果各成分阴性对照品相应保留时间无显著吸收,表明方法专属性良好。
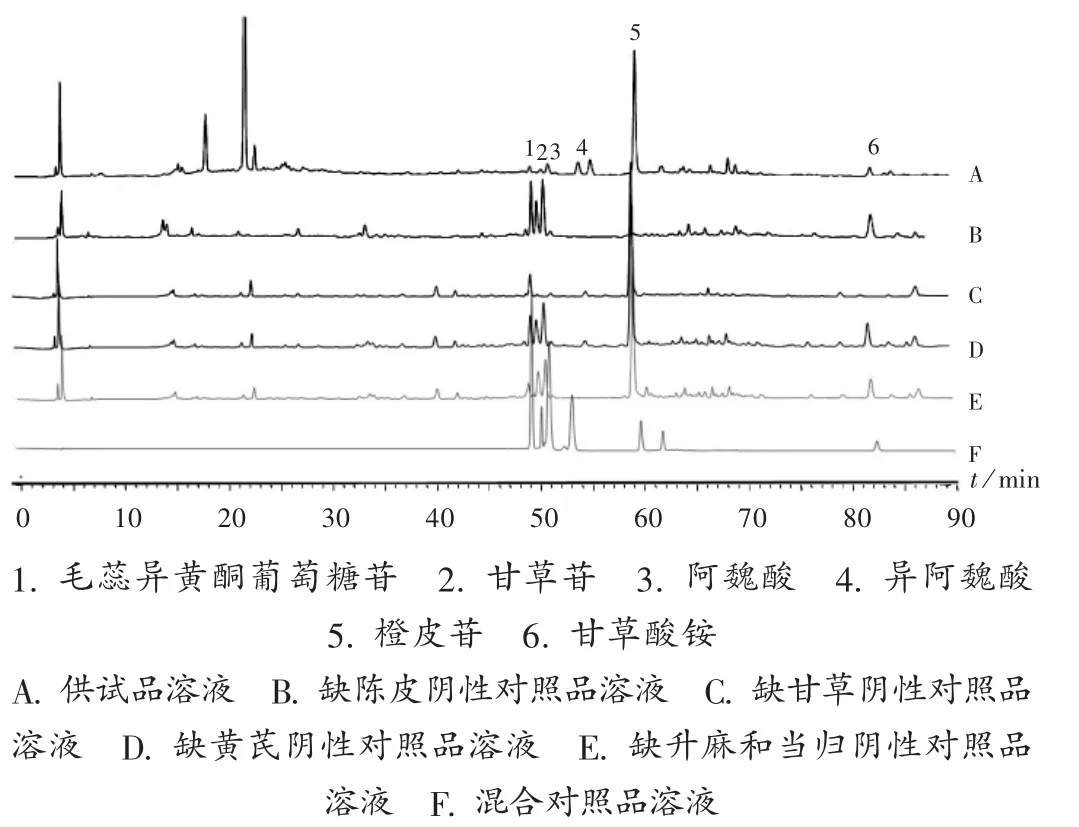
图2 专属性试验高效液相色谱图1.calycosin - 7 - O - β- D - glucoside 2.liquiritin 3.ferulic acid 4.isoferulic acid 5.hesperidin 6.ammonium glycyrrhizinateA.Test solution B.Negative reference solution lacking Citri Reticulatae Pericarpium C.Negative reference solution lacking Glycyrrhiza Radix et Rhizoma D.Negative reference solution lacking Astragali Radix E.Negative reference solution lacking Cimicifugae Rhizoma and Angelicae Sinensis Radix F.Mixed reference solutionFig.2 HPLC chromatograms of the specific test
线性关系考察:精密吸取2.2 项下系列混合对照品溶液,共7 份,按2.1 项下色谱条件进样分析,以各成分质量浓度(X,μg/mL)为横坐标、峰面积(Y)为纵坐标进行线性回归。结果见表2。
精密度试验:取2.2 项下混合对照品溶液,按2.1项下色谱条件连续进样测定6 次,记录峰面积。结果见表2,表明仪器精密度良好。
稳定性试验:取同一批(批号为15082417)样品,依法制备供试品溶液,按2.1 项下色谱条件分别于溶液制备后 0,4,8,12,18,24 h 时进样测定,并记录待测成分的峰面积。结果见表2,表明供试品溶液在24 h 内稳定性良好。
重复性试验:取同一批(批号为15082417)样品,依法制备供试品溶液,共6 份,按2.1 项下色谱条件进样分析,记录待测成分的峰面积。结果见表2,表明方法重复性良好。
加样回收试验:取同一批(批号为15082417)样品,粉碎,取粉末0.5g,精密称定,置具塞锥形瓶中,平行9 份,随机分为3 组,分别加入低、中、高3 种质量浓度的各成分混合对照品溶液适量,每组平行3 份,依法制备供试品溶液,按2.1 项下色谱条件进样测定,计算回收率。结果见表2。
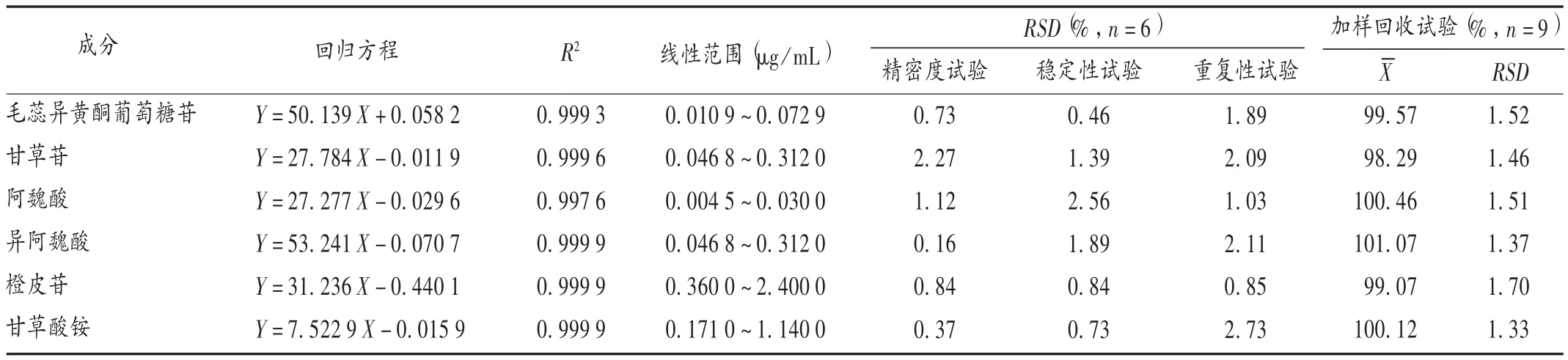
表2 补中益气丸中6 种成分方法学考察结果Tab.2 Results of the methodological investigation of six components in Buzhong Yiqi Pills
2.4 相对校正因子(RCF)确定
RCF 计算:取2.2 项下混合对照品溶液适量,以橙皮苷为内参物,在线性范围内按RCF 计算公式 fs/i=fs/ fi= (Ci× As)/(Cs× Ai)分别计算橙皮苷对毛蕊异黄酮葡萄糖苷、甘草苷、阿魏酸、异阿魏酸、甘草酸铵的RCF。式中,Cs 为内参物的质量浓度,As 为内参物的峰面积,Ci为待测组分的质量浓度,Ai为待测组分的峰面积[15]。结果见表 3。

表3 补中益气丸中5 种成分的相对校正因子(以橙皮苷为内参物)Tab.3 RCFs of five components in Buzhong Yiqi Pills(taking hesperidin as the internal reference)
RCF 的重复性:取2.2 项下混合对照品溶液适量,以橙皮苷为内参物,按2.1 项下色谱条件分别采用不同品牌(Dionex P680 型、Thermo LPG - 3400SDN 型)高效液相色谱仪和不同品牌色谱柱(Diamosil C18柱、Kromasil C18柱)测定,考察其对RCF 的影响。结果见表4。
2.5 色谱峰定位方法考察
取2.2 项下混合对照品溶液适量,以橙皮苷为内参物,按2.1 项下色谱条件测定,分别计算不同品牌(Dionex P680 型、Thermo LPG -3400SDN 型)高效液相色谱仪和不同品牌色谱柱(Diamosil C18柱、Kromasil C18柱)下其他5 种待测成分的相对保留时间差(ΔtR),即ΔtR待测成分/内参物= ΔtR内参物- ΔtR待测成分,以考察 ΔtR的重复性。结果见表4。
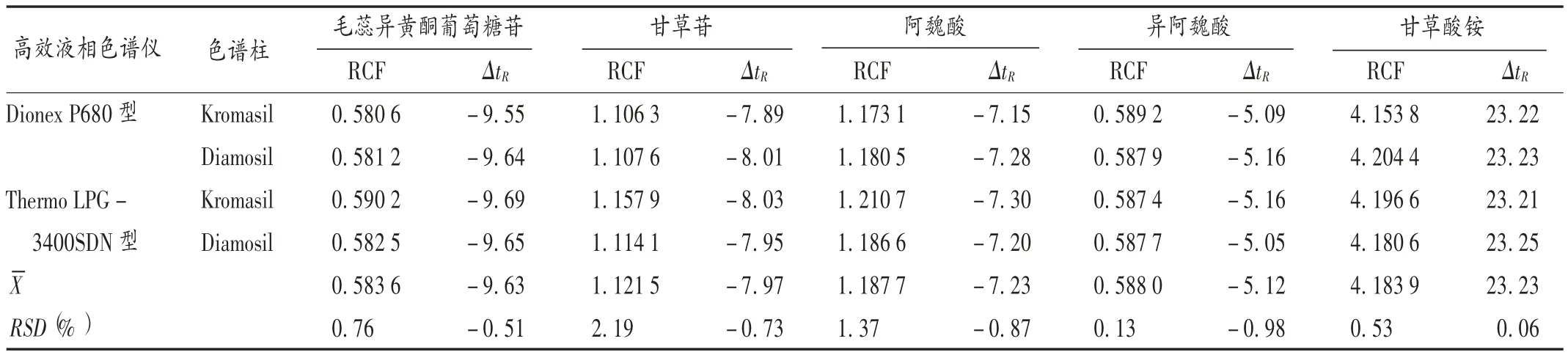
表4 不同仪器及色谱柱测定补中益气丸中5 种成分的相对校正因子及保留时间差(以橙皮苷为内参物)Tab.4 RCFs and retention time difference of Buzhong Yiqi Pills determined by different instruments and chromatographic columns (taking hesperidin as the internal reference)
2.6 QAMS 法与外标法定量测定结果比较
取12 批样品,按2.2 项下方法制备供试品溶液,按2.1 项下色谱条件进样测定,记录峰面积,分别采用QAMS 法和外标(ESM)法计算样品中毛蕊异黄酮葡萄糖苷、橙皮苷、甘草苷、甘草酸铵、阿魏酸、异阿魏酸的含量,对2 种方法的检测结果进行成组 t 检验,同时以相对误差 [ RE =(QAMS计算值-ESM实测值)/ESM实测值×100% ]对 2 种方法测定结果进行比较[16-18],详见表 5。结果显示,各阶段数据均服从正态分布,组间 P 值均大于 0.05,同时 2 种方法测得含量的|RE |均小于 5% ,表明2 种方法测得结果无显著差异,QAMS 法用于补中益气丸中6 种成分的含量测定可信度较好,可用于同时测定补中益气丸中多个组分的含量,可为全面评价补中益气丸的质量提供数据基础。

表5 QAMS 法与ESM 法测得补中益气丸中6 种成分的含量比较(n =2)Tab.5 Comparison of the content of six components in Buzhong Yiqi Pills determined by QAMS and ESM(n = 2)
3 讨论
3.1 内参物选择
参考文献[17-18],橙皮苷价廉易得,且其出峰附近干扰成分少,容易辨认,性质稳定。故以橙皮苷为内参物,采用QAMS 法对其他指标成分进行定量测定。
3.2 色谱峰定位方法选择
前期分别考察了 ΔtR、相对保留时间值和保留时间值用于待测色谱峰定位的稳定性,结果发现 ΔtR在不同仪器和色谱柱条件下波动较小。故选择耐用性更强的ΔtR法作为色谱峰最终定位标准。
3.3 色谱条件优化
参考文献[18-19],采用光电二极管阵列检测器在190 ~400 nm 波长范围内对混合对照品溶液进行扫描,分别考察6 种待测成分的紫外吸收情况。结果显示,于220 nm 波长处杂质峰较少,但甘草酸铵的色谱峰吸收不明显;于250 nm 波长处甘草苷和橙皮苷的色谱峰均有干扰;于285 nm 波长处毛蕊异黄酮葡萄糖苷的色谱峰有干扰且橙皮苷的色谱峰拖尾严重;于270 nm 波长处各待测成分均有较强吸收且峰形和分离度均好。故确定含量测定波长为270 nm。
考察了不同有机相(乙腈和甲醇)与水相(水和磷酸溶液)的流动相系统,并不断调整有机相-水相(磷酸溶液)的比例。结果显示,采用乙腈-0.1%磷酸水溶液进行梯度洗脱时,各组分的色谱峰响应最好且分离完全,基线平稳,重复性较好,保留时间较稳定。故最终确定流动相为乙腈-0.1%磷酸水溶液。
3.4 方法评价
中药成分复杂多样,在进行质量控制时需进行多组分测定,由于所需对照品数量众多且大多费用高昂,不利于广泛应用[20-22]。QAMS 法只需测定样品中单一成分即可实现其他多种成分的同步测定,适用于中药多组分的质量评价。补中益气丸中药味较多、成分复杂,本试验选择了毛蕊异黄酮葡萄糖苷、甘草苷、甘草酸铵、阿魏酸、异阿魏酸、橙皮苷6 种具有代表性的指标性成分进行考察。结果显示,以橙皮苷为内参物,不同品牌色谱仪和色谱柱对其余5 种成分的RCF 的影响较小,重复性和稳定性均良好,故建立了上述6 种指标性成分含量测定的QAMS 法。该方法与ESM 法测得的含量无显著差异,可在仅有1 个对照品时实现同步测定补中益气丸中的多种指标性成分,克服了对照品短缺及检测成本高的困难,为补中益气丸的质量控制研究提供了参考。
