ldentification and functional prediction of long intergenic noncoding RNAs in fetal porcine longissimus dorsi muscle
2021-12-14
College of Life Sciences, Xinyang Normal University, Xinyang 464000, P.R.China
Abstract Pigs are globally farmed animals which provide protein for human consumption in the form of skeletal muscle. To better understand the function of long intergenic noncoding RNAs (lincRNAs) in porcine skeletal muscle growth and development,we collected RNA-seq data from porcine longissimus dorsi muscle (LDM) during embryonic development. We identified a total of 739 lincRNA transcripts,which were distributed on all chromosomes except the chromosome Y,and analyzed their molecular characteristics. Compared to protein-coding genes,lincRNAs showed shorter transcripts,longer exons,fewer exons and higher tissue specificity. In addition,the abundance of lincRNAs in five embryonic development stages were analyzed and 45 differentially expressed lincRNAs were screened,three of which were highly expressed in LDM during porcine embryonic development. Finally,we predicted the potential target genes and functions of the lincRNAs,and identified 1 537 cis-target genes and 8 571 trans-target genes. Furthermore,we identified two key candidate lincRNAs involved in muscle development,XLOC_024652 and XLOC_001832,for post-trial validation. Our results provide a genome-wide resource of lincRNAs which are potentially involved in porcine embryonic skeletal muscle development and lay a foundation for the further study of their functions.
Keywords:lincRNAs,pig,skeletal muscle,embryo,potential target genes
1.lntroduction
Long intergenic noncoding RNAs (lincRNAs) are a class of noncoding RNAs which are greater than 200 bp in length and have limited protein-coding potential (Talyanet al.2018;Zouet al.2018). Genomic studies indicate that lincRNAs are ubiquitous in mammals and they participate in various biological processes,such as tumorigenesis,embryonic development,metabolism and skeletal muscle development,through either transcriptional,post-transcriptional or epigenetic regulation (Li Cet al.2018). The development of next generation sequencing (NGS) technology has greatly facilitated the identification of skeletal muscle lincRNAs,especially in human and mouse (Li Yet al.2018; Chenet al.2019). Therefore,the identification of lincRNAs and analysis of their molecular regulatory mechanisms in livestock will provide a theoretical basis for the genetic improvement of vital economic traits.
Susscrofais not only a major meat source but also a biomedical model for various human disease because of the extensive similarities between the body size and physiological features of humans and pigs (Tanget al.2007; Zouet al.2018). The modern pig industry focuses its research efforts on growth rate and high meat quality(Stachowiak and Flisikowski 2019). Meat quality is closely related to the amount of muscle fibers before birth,so fetal muscle development is important for subsequent muscle growth and meat quality (Wigmore and Stickland 1983; Tanget al.2007; Liuet al. 2018). Skeletal muscle development is a complex multistep biological process which is regulated by multiple genes,transcription factors,growth factors,epigenetic regulators and noncoding RNAs (Chenet al.2019). Although some key genes and regulators with important roles in skeletal muscle development have been discovered,such asMyoDandPHKG1,existing studies have mainly focused on protein-coding genes (Xionget al.2011; Maet al.2014). The role of lincRNAs in porcine fetal skeletal muscle development is still unclear.
Longissimus dorsimuscle (LDM) is closely associated with important economic performance characteristics of livestock carcass production,but few reports on lincRNAs in the LDM of pig fetuses exist. Therefore,we hypothesized that some lincRNAs related to the skeletal muscle development in pig fetuses probably have yet to be discovered. The systematic identification and analysis of the lincRNAs in LDM can lead to the discovery of a series of lincRNAs related to skeletal muscle development.Therefore,the aim of this study was to identify and analyze the lincRNAs in the LDM of pig fetuses in order to test the above hypothesis and provide valuable new material for future improvements in commercial pig breeding.
2.Materials and methods
2.1.Data acquisition
PolyA+RNA-seq data from pig LDM in various embryonic development (ED) stages were obtained from the research of Liuet al.(2018) and were downloaded from the NCBI SRA website (https://www.ncbi.nlm.nih.gov/Traces/study/?acc=SRP066398&o=acc_s%3Aa) using the project number SRP066398. The accession numbers and detailed information of the pig embryonic RNA-seq(including five embryonic stages with one repeat in each of two breeds,Tongcheng pig and Yorkshire pig) are given in Appendix A. From assessing tissue specificity,RNA-seq data from eight pig tissues were collected from the NCBI SRA website (https://www.ncbi.nlm.nih.gov/Traces/study/?acc=ERP009821&o=acc_s%3Aa),and the accession numbers and detailed information are listed in Appendix B.
2.2.Publicly available annotations
The genome reference sequence and annotation (version 11.1)were downloaded from the NCBI Database. NCBI genome annotation was utilized to characterize the novel lincRNAs,as compared to all annotated lincRNAs and partial proteincoding transcripts.
2.3.RNA-seq data cleaning,mapping and assembly
Raw RNA-seq sequence reads were first subjected to quality control using FastQC (version 0.11.5),and were then cleaned according to the following criteria using Trimmomatic (version 0.33) (Bolgeret al.2014):(1) remove the adaptor; (2) cut once based on the average quality within the window that falls below 15 when performing a sliding window trimming with a 4 base window size; (3) drop the read if it is less than 36 bp.
The cleaned RNA-seq datasets were then aligned with theSusscrofa11.1 genome sequence using Tophat Software (version 2.1.1) and combined with the genome annotation using the default parameters (Pollieret al.2013).
For each embryonic stage,mapped reads from Tophat were assembled for each sample separately by Cufflinks(version 2.2.1) (Trapnellet al.2010). Cufflinks uses a probabilistic model to assemble and quantify the expression levels of a minimal set of isoforms,and it provides a maximum likelihood explanation of the expression data for given loci. Then all assembled transcript files of the embryonic stages were merged into a unique transcriptome using Cuffmerge which was obtained from the Cufflinks package.
2.4.Expression analysis
The sam file was transformed from the bam file,which was produced by Tophat using samtools (version 0.1.18) (Liet al.2009),then the unique mapping reads were obtained using samtools. The number of reads which mapped to the lincRNA was calculated by htseq-count with the parameter-m union -s reverse (Anderset al.2015). Then we analyzed the differential expression of lincRNA pairwise comparisons using DEGseq based on the R language (Wanget al.2010)and Gfold v1.1.4 (Fenget al.2012). For the results of Gfold,lincRNA genes with an RPKM value greater than 0.3 in at least one sample and a Gfold value greater than 1 or less than -1 were considered as differentially expressed lincRNA genes (DELs). For DEGseq,lincRNA genes with mapping reads greater than 10 in at least one sample,log2(fold change) greater than 1 or less than -1 and with a signature that is TRUE were considered as DELs. Finally,the lincRNA genes which were determined to be differentially expressed using both methods were considered as the DELs.
2.5.The pipeline for identifying lincRNAs
To identify novel lincRNAs from the pig embryonic LDM transcriptome,we designed an analysis pipeline which identifies the lincRNAs using the result of Cuffmerge through the following steps:(1) only transcripts placed in the ‘u’ category by Cuffmerge which indicates intergenic transcripts were retained; (2) transcripts with a single exon or less than 200 bp in length were removed; (3) the coding potential calculator (CPC) tool (Konget al.2007)was used to assess the coding potential of transcripts in both strands,and only transcripts with CPC value <0 for both strands were retained; (4) transcripts that contained a known protein domain were filtered. To accomplish this,we translated transcript sequences into six possible protein sequences using Transeq (http://www.ebi.ac.uk/Tools/st/emboss_transeq/),and then transcripts with any possible protein sequence which was significant (E-value<1e-5) hits in the Pfam Database (http://pfam.xfam.org/search) were filtered; and (5) to minimize false positives,we removed transcripts for which FPKM was less than 0.3 in all samples(Fig.1).
2.6.Tissue specificity analysis
The tissue specificity index (τ) (Yanaiet al.2005; Liao and Liao 2006; Xianget al.2010),which is used to measure the tissue specificity of a gene,is defined as:

whereNis the number of tissues examined (equal to eight here),X(i,max)is the highest number of reads mapped to geneiacross the N tissues,andX(i,j)is the number of reads mapped to geneiin tissuej. Theτvalues range from 0 to 1,which 1 indicates one tissue specificity and 0 indicates an internal reference gene.
2.7.Prediction and functional analysis of potential target genes of the lincRNAs
Based on the position information of the lincRNAs,we defined the protein-coding genes within the range of 100 kb around a lincRNA as thecis-target genes of that lincRNA.BEDtools (version 2.25.0) was used to identify thecis-target genes of linRNAs according to the annotation of lincRNAs and the pig reference genome (Quinlan and Hall 2010). The correlations of expression of a lincRNA and itscis-target genes were calculated using R scripts based on FPKM.
In addition,we defined protein-coding genes that are strongly correlated with the expression of lincRNAs astranstarget genes of those lincRNAs. To identify the potential target genes (PTGs) that were regulated by lincRNAs astrans-targets,the expression levels of candidate lincRNAs and the protein-coding genes were used,and Pearson’s correlation coefficients between each pair of lincRNAs and the protein-coding genes were calculated (r>0.95 orr<-0.95 andP<0.01).
Functional enrichment analyses of these PTGs were generated using DAVID Online Software (Huanget al.2009).Because of the limited annotation of the porcine genome,all PTGs were first converted into their human homologous genes using BIOMART in Ensembl. Only significantly enriched GO terms and KEGG pathways were reported(i.e.,those with Benjamini-Hochberg correctedP<0.05).
3.Results
3.1.ldentification and characterization of porcine fetal LDM lincRNAs
In order to systematically identify and analyze the lincRNAs associated with skeletal muscle development in embryonic pigs,the published LDM RNA-seq data for five embryonic stages (E40,E55,E63,E70 and E90) of Yorkshire and Rongchang pigs (Liuet al.2018) were used to identify lincRNAs following a procedural pipeline (Fig.1). A total of 739 candidate lincRNA transcripts were identified,which were encoded by 538 lincRNA genes. These genes are distributed among all chromosomes except chromosome Y (chrY). Notably,the lincRNA genes are enriched in chr1,chr3,chr6,and chr13 (Fig.2-A; Appendix C).
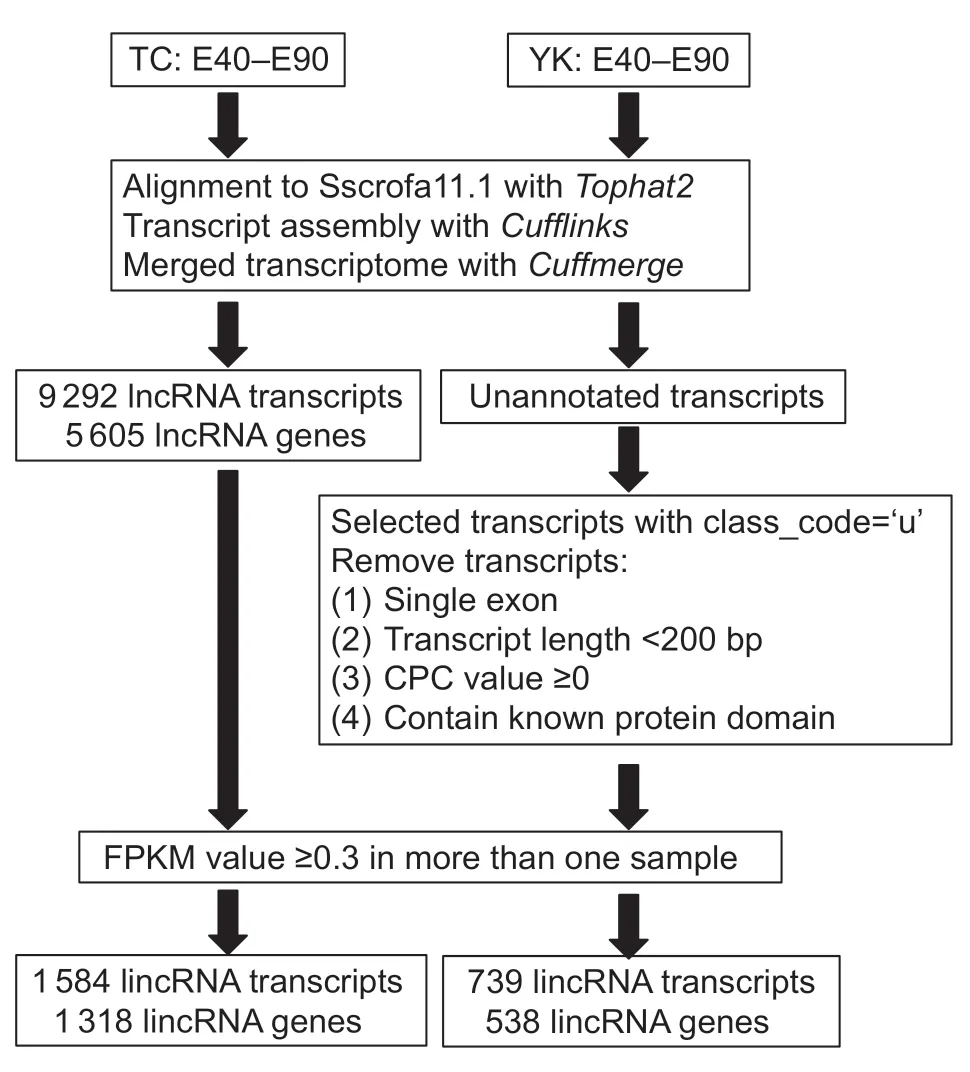
Fig.1 The pipeline for novel long intergenic noncoding RNAs(lincRNAs) in this study.
We then compared the porcine fetal LDM lincRNAs with pig protein-coding genes (Fig.2-B-D). The porcine fetal LDM lincRNAs had shorter transcript lengths and longer exon lengths than the known lncRNA genes and protein-coding genes (Ks.test,P<2.2e-16) (Fig.2-B and C).Furthermore,the candidate lincRNA genes showed fewer exons than known lncRNA genes and protein-coding genes(Ks.test,P<2.2e-16) (Fig.2-D). These characteristics of the porcine fetal LDM lincRNA genes are consistent with previous studies (Li Cet al.2018; Zouet al.2018).
3.2.Tissue-specific analysis of lincRNAs
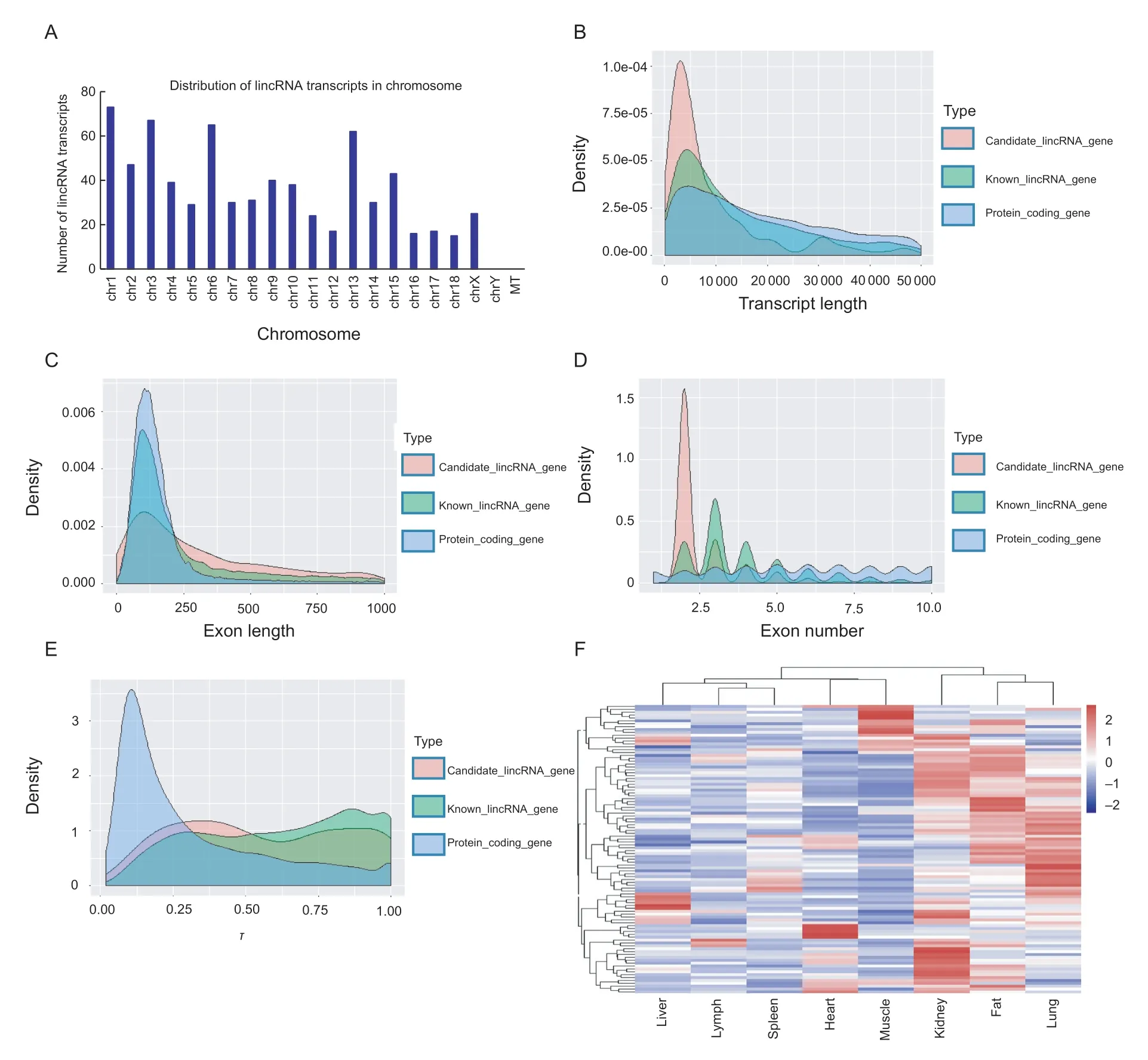
Fig.2 The characteristics analysis of novel long intergenic noncoding RNAs (lincRNAs). A,distribution of identified lincRNA transcript in chromosome. B,C,and D,comparisons of transcript length,exon length,and exon numbers among novel lincRNA genes,known lncRNA genes and protein-coding genes,respectively. E,tissue-specific analysis of novel lincRNA genes,known lncRNA genes and protein-coding genes. τ,tissue specificity index. F,the expression heat map of lincRNA genes,which mapping reads have more than 10 reads in eight tissues.
To gain a comprehensive understanding of tissue-specific expression profiles between the porcine fetal LDM lincRNA genes and protein-coding genes,we analyzed the specificity of their tissue expression using published RNA-seq data from eight types of tissues:heart,liver,spleen,lung,kidney,muscle,fat and lymph gland (Appendix B). The mean τ values of lincRNA genes,known lncRNA genes and protein-coding genes were 0.56,0.62 and 0.32,respectively.Compared with protein-coding genes,the lincRNA genes and known lncRNA genes exhibit higher tissue specificities(Wilcox.test,P<2.2e-16) (Fig.2-E). To characterize lincRNA gene expression features,we selected the lincRNA genes that had more than 10 reads in the eight tissues. We found that most of the lincRNA genes were highly expressed specifically in muscle,fat,kidney and lung (Fig.2-F). These results are consistent with those of our previous study (Li Cet al.2018).
3.3. Differential expression analysis of lincRNAs
In order to explore the expression levels of our identified lincRNAs,we first compared the expression level between porcine fetal LDM lincRNAs and protein-coding genes using porcine fetal LDM RNA-seq data and found that the putative lincRNAs have significantly lower expression levels than the protein-coding genes (Wilcox.test,P<2.2e-16) (Fig.2-A;Appendix D). These results are consistent with previous results and show that lincRNAs have lower expression in the entire genome (Zouet al.2018).
Skeletal muscle development is affected by the number of muscle fibers,which is determined before birth (Wigmore and Stickland 1983; Tanget al.2007; Zhaoet al.2015;Liuet al.2018),so the stage-specific lincRNAs may play a critical role in skeletal muscle development. Here,we analyzed the expression of lincRNAs using RNA-seq data from Rongchang pig and Yorkshire pig at five embryonic stages (Appendix A). By pairwise comparison (E40vs.E55,E55vs.E63,E63vs.E70,E70vs.E90),we identified 123 DELs in Tongcheng pig and 97 DELs in Yorkshire pig during skeletal muscle development,and the DELs of each comparison group are shown in Appendix E. Interestingly,many more DELs have higher expression levels in E55 when compared to E40 or E63,especially in Yorkshire pig(Appendix E). Secondary fibers first appear at E54 (Wigmore and Stickland 1983),so the DELs with high expression levels at that point may be involved in the formation of secondary fibers. In addition,more DELs have lower expression levels in E90 when compared to E70 (Appendix E). The number of primary fibers begins to decline after E80,while the formation of secondary fibers ends at E90 (Wigmore and Stickland 1983; Tanget al.2007),so the DELs which are more highly expressed in E70 than E90 may play roles in the formation of primary and secondary fibers. Combining the DELs of both Tongcheng and Yorkshire pigs,a total of 45 DELs were identified (Fig.3-B; Appendix E). Among these 45 DELs,three lincRNA genes (XLOC_029943,XLOC_024652andXLOC_034153) were more highly expressed in LDM during porcine fetal development,so they may play crucial roles in pig skeletal muscle development.
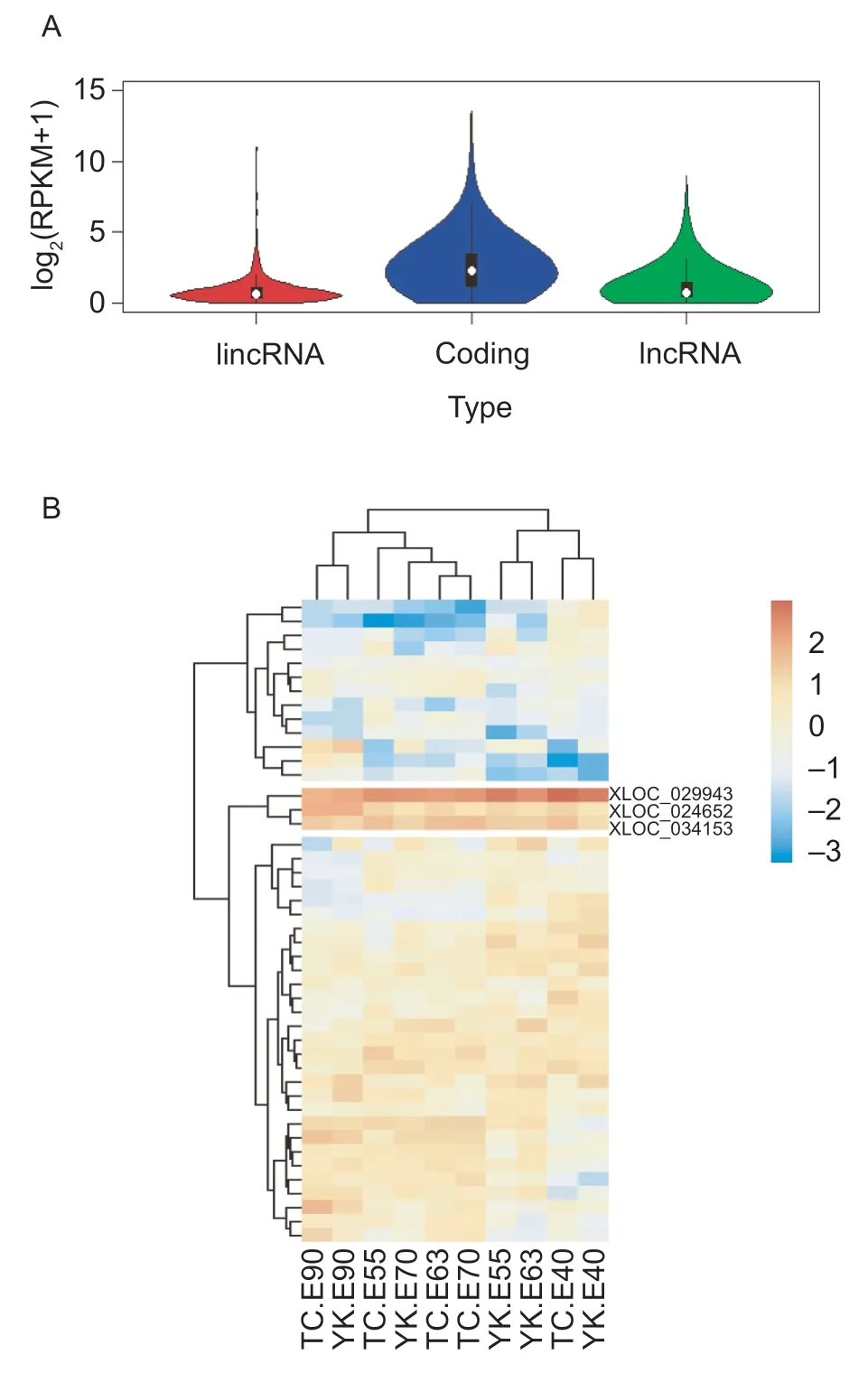
Fig.3 The expression analysis of long intergenic noncoding RNAs (lincRNAs) genes. A,comparison of expression level among novel lincRNA genes (lincRNA),known lncRNA genes(lncRNA) and protein-coding genes (coding). B,the expression heat map of differentially expressed lincRNA genes (DELs). The expression level was calculated by log2(RPKM+1).
3.4.Prediction of potential target genes and functional prediction of lincRNAs
It is known that lincRNAs can regulate the expression of potential target genes through eithercis-ortrans-regulation.Furthermore,the potential target genes of lincRNA genes can be predicted according to the positional relationship and expression correlations between the coding genes and lincRNA genes (Bakhtiarizadeh and Salami 2019).
In order to predict the potentialcis-target genes of our lincRNAs,we used a threshold distance of 100 kb between the lincRNAs and genes,and predicted 1 537cis-target genes across all the lincRNAs (Appendix F). Thirty-one of the 45 DELs were detected close to 93 mRNAs,in which the targets ofXLOC_024652andXLOC_034153wereNRP1andPPP1R3B,respectively (Appendix G). Since the annotation information of pigs is incomplete,we screened human homologous genes of thesecis-target genes and then performed functional annotation and KEGG pathway analysis. By performing GO analysis,we found that thesecis-target genes participate in many biological processes,such as transcription regulation,the apoptotic process and protein phosphorylation (Fig.4-A). In addition,KEGG analysis showed that thesecis-target genes were mainly enriched in the signaling pathway,fatty acid degradation and other pathways (Fig.4-B). Among them,NRP1participates in multiple biological processes,such as protein binding,metal ion binding and the semaphorin-plexin signaling pathway. These results indicated that these candidate lincRNAs may regulate theircis-target genes by transcriptional regulation,protein phosphorylation or other epigenetics patterns,and further participate in multiple signaling pathways.
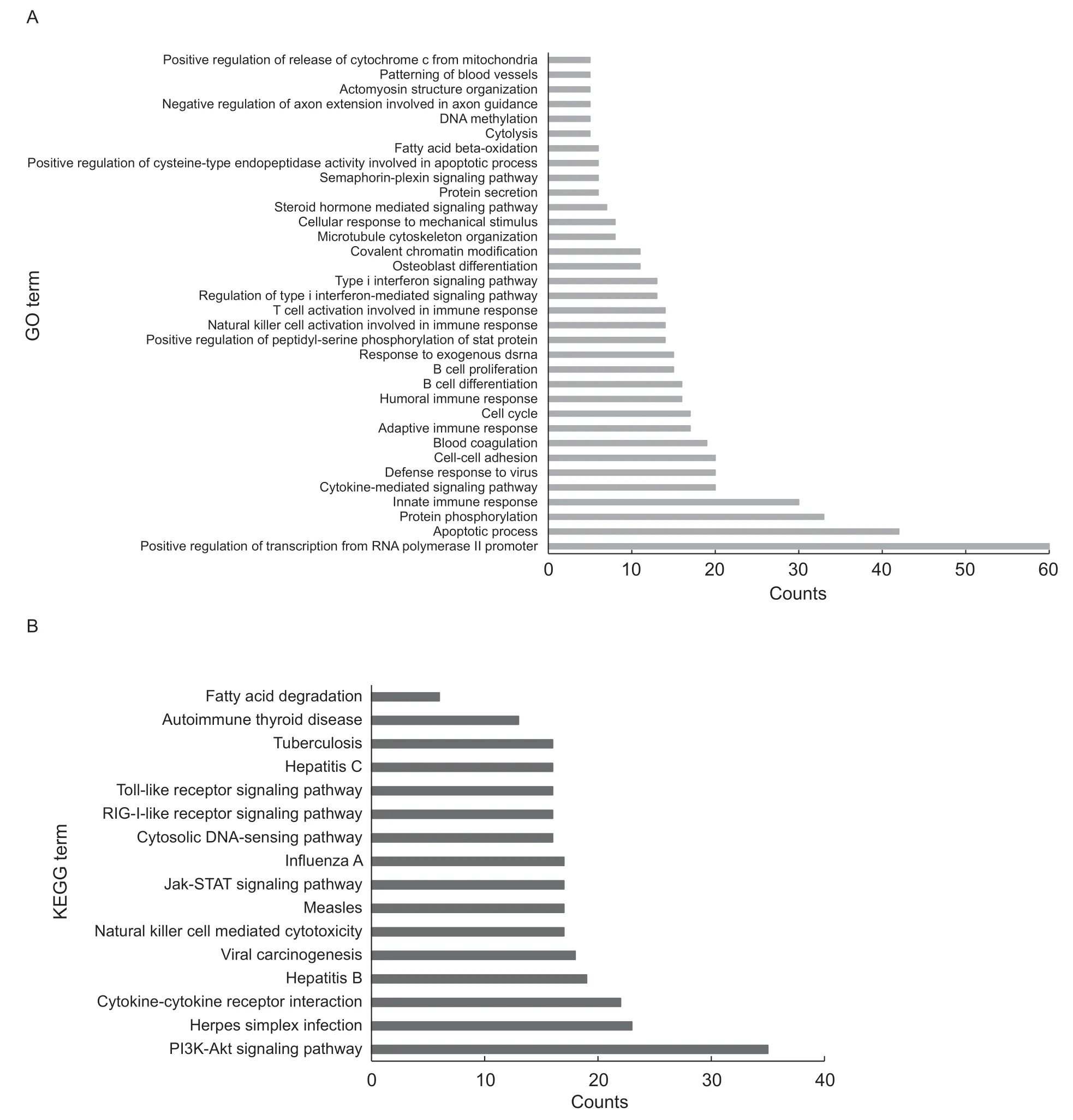
Fig.4 GO (A) and KEGG (B) analyses of cis-target genes of candidate long intergenic noncoding RNAs (lincRNAs) genes.
Next,we predicted thetrans-regulatory target genes of lincRNAs by constructing an mRNA-lincRNA co-expression network based on values ofr>0.95 orr<0.95 andP<0.01.We identified 8 571 significant correlations including 3 919trans-target genes,among which 4 852 were positive correlations and 3 719 were negative correlations (Fig.5;Appendix H). Compared with the results of thecis-target gene prediction,we found that thecis-target genes andtrans-target genes of two lincRNAs were identical(XLOC_000633:target geneMEF2AandXLOC_001832:target geneLOC102163334),in whichXLOC_001832is a DEL in LDM during pig embryonic development (Fig.3-B).Another interesting gene isZNF609,which is thetranstarget gene of DELXLOC_034153,and it participates in multiple muscle developmental biology processes,such as muscle organ development and regulation of myoblast proliferation (Legniniet al.2017). To predict the functions of the candidate lincRNAs,we performed functional enrichment analysis based on their relatedtrans-target mRNAs. Fiftythree GO terms and 16 KEGG pathways were significantly enriched,such as the Notch signaling pathway,muscle contraction,positive regulation of the gene expression and fatty acid metabolism pathway,the insulin resistance pathway,and others (Fig.6).
4.Discussion
With the development of high-throughput sequencing technology,a large number of lncRNAs have been identified,especially in human and mice (Zouet al.2018). However,fewer lncRNAs have been found in pigs (Weikardet al.2017).The results of our current study support our hypothesis that some undiscovered lincRNAs might be related to the skeletal muscle development in pig fetuses. In the present study,a number of novel lincRNAs were identified in the LDM of pig fetuses,and some of these novel lincRNAs were found to be associated with the skeletal muscle development of pig fetuses. Therefore,this study provides valuable materials for the genetics and breeding of pigs.
In the current study,739 putative lincRNA transcripts were identified,which are distributed on all chromosomes,except the chromosome Y through strict screening criteria.Our results will contribute to future studies on pig lincRNAs,particularly those expressed in the porcine fetal LDM,by adding a novel batch of muscle lincRNAs to the known pig lincRNA resources. Molecular characteristic showed that the putative lincRNAs have shorter transcripts,longer exons and fewer exons compared to protein-coding genes,which is similar to the lincRNAs reported in previous studies (Liet al.2016; Tanget al.2017; Zouet al.2018; Li Xet al.2019).
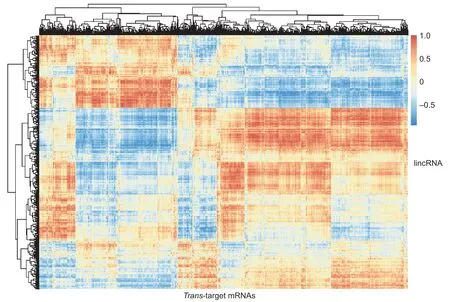
Fig.5 The correlation relationship between long intergenic noncoding RNAs (lincRNAs) and their trans-target mRNAs.
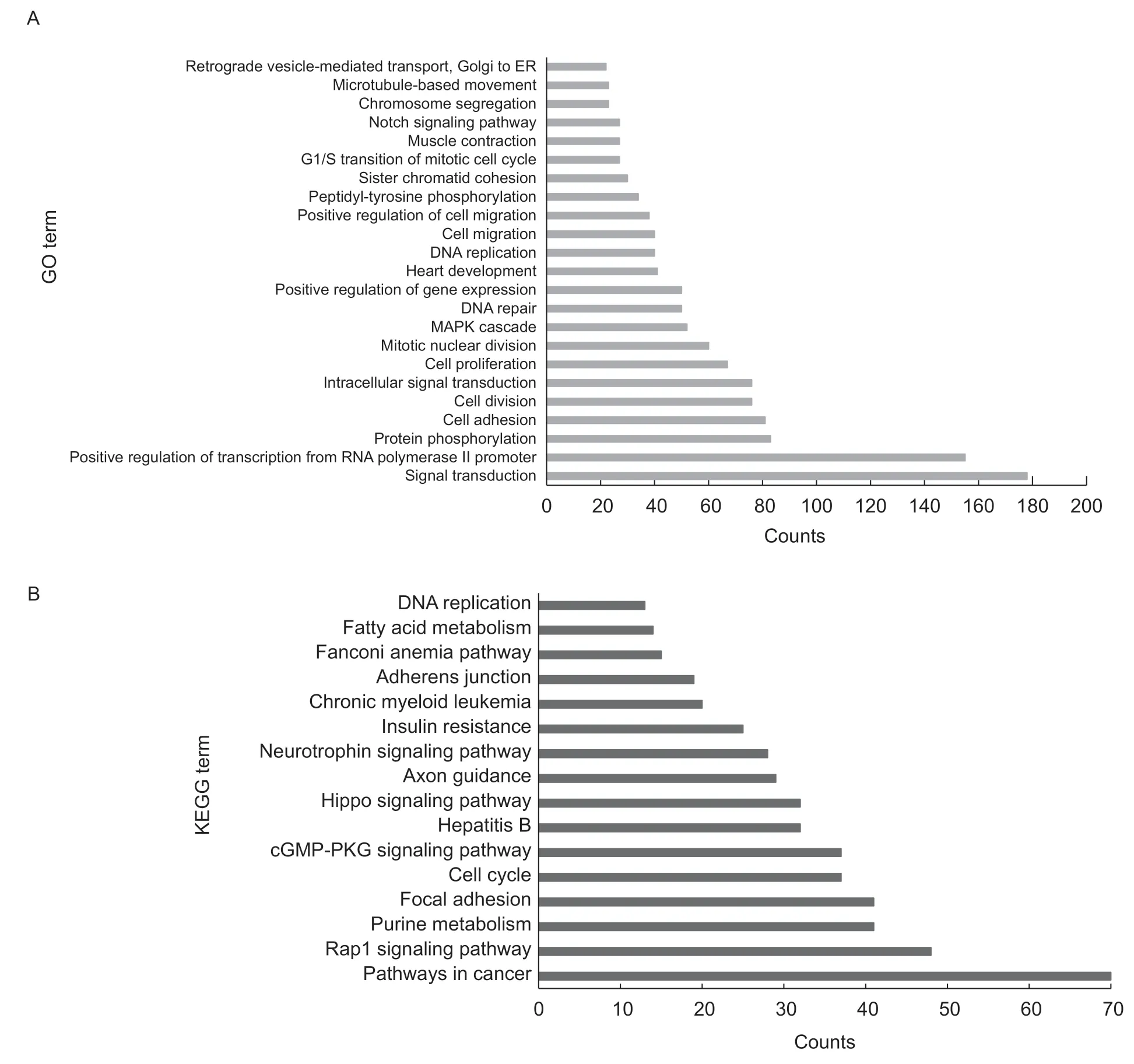
Fig.6 GO (A) and KEGG (B) analyses of trans-target genes of candidate long intergenic noncoding RNAs (lincRNAs).
Tissue-specific analysis showed that the putative lincRNAs exhibit high tissue specificity,which is also consistent with previous studies (Pauliet al.2012; Tanget al.2017). More putative lincRNAs were specifically highly expressed in muscle,fat,kidney and lung,so some of these putative lincRNAs play an important role in muscle development during pig embryogenesis. While muscle is an important metabolic tissue involved in a variety of biological processes,here we focus on the lincRNAs involved in muscle development in the pig embryo.
In order to screen for lincRNAs that play major roles in pig muscle development,we found 45 DELs in pig LDM during embryonic development. Among these DELs,three lincRNAs (XLOC_029943,XLOC_024652,andXLOC_034153) were highly expressed in LDM during pig embryonic development. We hypothesized that these three lincRNAs may play a vital role in pig muscle development,which may affect the muscle metabolism,meat quality and other characteristics of the pigs,although this remains to be verified by further study.
Compared with the discovery and characterization of lincRNAs,the study of their biological functions is a more challenging task. Previous studies have shown that lincRNAs preferentially exertcis-regulating effects on mRNAs located in very close proximity (Jandura and Krause 2017). Therefore,the functions of these putative lincRNAs can be inferred by the functions of the protein-coding genes located within 100 kb upstream and downstream of the lincRNAs. In our study,1 537 protein-coding genes were predicted to be located within around 100 kb of all the putative lincRNAs,of which 93 mRNAs were close to 31 DELs. Functional enrichment analysis showed that the adjacent mRNAs were mainly concentrated in transcription regulation,the apoptotic process,protein phosphorylation and fatty acid degradation. Based on these results,we predict that these putative lincRNAs may regulate their adjacent protein-coding genes which participate in fatty acid degradation and other biological processes through transcription regulation,protein phosphorylation or other epigenetic regulation mechanisms,and thereby regulate muscle development. Interestingly,NRP1,one of thesecis-target genes,is located nearXLOC_024652,which is a differentially expressed lincRNA,and enriched in the terms of protein binding,metal ion binding,and the semaphorin-plexin signaling pathway. Previous research showed thatNRP1plays critical roles in the growth of new blood vessels and heart regeneration (Hwanget al.2019;Loweet al.2019). Moreover,Li Het al.(2019) showed thatmicroRNA-320targetNRP1inhibits the proliferation and migration of vascular smooth muscle cells and neointimal formation. Therefore,XLOC_024652may targetNRP1and then participates in pig muscle development,but this conjecture remains to be tested.
Another common method for predicting target genes of lincRNAs is the use of coexpression analysis to predict which mRNAs have similar expression patterns as thetranstarget genes of lincRNAs. This method enables lincRNAs to regulate mRNAs distant from their transcription sites.In this study,8 571 significant correlations were identified between lincRNAs and protein-coding genes,which included 4 852 positive correlations and 3 719 negative correlations.Thesetrans-target genes are mainly enriched in the Notch signaling pathway,muscle contraction,positive regulation of gene expression,fatty acid metabolism and insulin resistance. Through these results,we speculate that these lincRNAs may be involved in diverse biological processes such as muscle development,fatty acid metabolism and insulin resistance by regulating the expression of thesetrans-target genes. Interestingly,ZNF609,thetrans-target gene of DELXLOC_034153,is enriched in multiple muscle development processes,such as muscle organ development and regulation of myoblast proliferation. Furthermore,circ-ZNF609,which is produced byZNF609,also has an essential function in myogenesis (Legniniet al.2017;Rossiet al.2019; Voellenkleet al.2019). Therefore,we predict thatXLOC_034153may regulate theZNF609ring to formcirc-ZNF609through epigenetic regulation,and thus ultimately plays an important role in myogenesis.
5.Conclusion
We identified a number of lincRNAs associated with muscle development during pig embryo development and analyzed the molecular characteristics of these putative lincRNAs.Functional analysis showed that these putative lincRNAs mainly participate in transcriptional regulation,muscle development,fatty acid metabolism and other biological processes which are associated with muscle development.In addition,we found two interesting lincRNAs which may have critical function in myogenesis. These results improve our understanding of lincRNAs in pig muscle development and provide valuable clues for future research.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (31601167,31972537 and U1204326),the Natural Science Foundation of Henan Province,China (182300410027),the Central Plains Technological Innovation Leading Talents Project of Henan Province,China (194200510022),and the Nanhu Scholars Program of Xinyang Normal University,China.
Appendicesassociated with this paper can be available on http://www.ChinaAgriSci.com/V2/En/appendix.htm
杂志排行

Journal of Integrative Agriculture的其它文章
- lnfectious disease-resistant pigs:Will they fly?
- Crop photosynthetic response to light quality and light intensity
- Alginate oligosaccharides preparation,biological activities and their application in livestock and poultry
- Genome-wide pedigree analysis of elite rice Shuhui 527 reveals key regions for breeding
- TaSnRK2.4 is a vital regulator in control of thousand-kernel weight and response to abiotic stress in wheat
- Development and characterization of new allohexaploid resistant to web blotch in peanut