INRA717-1B4杂交杨TAC基因CRISPR/Cas9敲除载体的构建及编辑效率检测
2021-12-11郝璞薄高峰刘璐白鹤鸣吴凤霞周星雨杨海峰
郝璞,薄高峰,刘璐,白鹤鸣,吴凤霞,周星雨,杨海峰
(内蒙古农业大学林学院,内蒙古 呼和浩特 010018)
CRISPR/Cas9系统作为近年来被广泛开发应用的基因编辑工具,主要由核酸内切酶Cas9和一个单分子向导RNA(single guide RNA,sgRNA)组成,通过RNA与目标DNA的互补配对,Cas9蛋白在目标序列进行特异切割,造成DNA双链断裂,通过激活细胞自身修复机制——非同源末端连接(NHEJ)或同源重组(HR)来实现基因敲除、插入以及染色体重组等,最终导致随机突变的产生[1]。CRISPR/Cas9系统最早应用于动物和微生物中,后证实在水稻(Oryza sativa)、小麦(Triticum aestivum)、拟南芥(Arabidopsis thaliana)、烟草(Nicotiana benthamiana)、高粱(Sorghum bicolor)、玉米(Zea mays)等植物中也能实现定点编辑[2-8];之后宋时奎等[9]整理了CRISPR/Cas9系统在单子叶和双子叶植物中基因组定点编辑的案例,得出该系统在植物基因组进行定点编辑的可行性,特别是由于嵌合sgRNA的成功设计,使其应用更加简单方便。此后CRISPR/Cas9被应用于各个领域,在动植物基因功能验证、农作物性状改良以及人类疾病治疗方面展现出了巨大的发展前景[10,11]。CRISPR/Cas9系统的出现是分子育种学上革命性的技术突破,也是现代基因组学靶向修饰工作的巨大进步[12]。
INRA717-1B4杂交杨(Populus tremula L.×P.alba L.),属白杨派落叶乔木,具有基因组小、实验特性优良、易转化、生长繁殖迅速等特点[13]。作为木本植物的模式物种,CRISPR/Cas9系统在杨树中实现了广泛应用。Fan等[14]利用CRISPR/Cas9系统完成了杨树内源基因定点突变,成功敲除毛白杨(P.tomentosa Carr.)PDS基因(phytoene desaturase gene),导致杨树叶片白化。Zhou等[15]利用CRISPR/Cas9技术成功编辑717-1B4杨的4CL1和4CL2基因(4-coumarate:CoA ligase gene)。Bruegmann等[16]针对717-1B4杨选取了12个基因,包括调控开花时间基因SOC1、FUL及其同源基因、4个NFP基因和TOZ19基因,转化获得15个抗性株系,经PCR鉴定均为阳性植株。本研究利用CRSPR/Cas9系统对TAC基因进行敲除,以期为TAC在717-1B4杨的基因功能验证提供研究依据,并为其他作物TAC同源基因的功能验证提供材料。
1 材料与方法
1.1 试验材料
INRA717-1B4杂交杨(Populus tremula L.×P.alba L.,简称717-1B4杨),取自内蒙古农业大学林学院教研室;717-1B4杨全基因组数据信息源于717-1B4杨基因组数据库(aspendb.uga.edu/databases/spta-717-genome);入 门 载体pEn-Chimera、表达载体pDE-CAS9-NPTⅡ,均由美国农业部林务局西南太平洋工作站林木遗传所赠送;大肠杆菌DH5α感受态细胞、DB3.1感受态细胞,购自北京全式金生物技术有限公司;农杆菌GV3101感受态细胞,购自北京博迈德生物技术有限公司。
1.2 入门载体的构建
1.2.1 717-1B4杨TAC基因靶位点序列的设计 CRISPR/Cas9系统的切割过程主要凭借核糖核蛋白复合物crRNA和Cas蛋白,扫描外源序列的protospacer以及PAM,PAM序列不显现在靶点序列里。选择靶点时要尽量保证其特异性,从而降低脱靶概率。靶点要求长度为20 bp,靶位点特异性高,不要出现连续多个A或者T,3′端要有PAM序列,引物不显示PAM序列。在717-1B4杨中TAC基因的第一个旁系同源基因(Potri.002G175300)和第二个旁系同源基因(Potri.014G102600)筛选外显子序列,评估靶点的脱靶效应及GC含量,GC含量保证在40%~70%之间,设计两个靶位点sgRNA,分别命名为pDe-TAC1-1和pDe-TAC2-1。利用717-1B4杨基因组数据库及推荐的靶位点(spta717grnas.csv)结合毛果杨等相关基因组进行靶点比对,确保靶点的特异性。根据靶点合成相应引物(表1),由中美泰和生物技术(北京)有限公司合成,纯化级别为PAGE。

表1 本研究所用引物
1.2.2 pEn-Chimera入门载体的构建及转化利用限制性内切酶BbsⅠ将pEn-Chimera载体质粒进行酶切,酶切体系(20μL):pEn-Chimera 10μL、10×NEB buffer 2μL、NEB BbsⅠ1μL、ddH2O 7μL。37℃孵育1 h,孵育后的产物70℃反应15 min,使BbsⅠ失活。线性化后采用1%琼脂糖凝胶电泳检测反应产物,使用试剂盒(EasyPure®Quick Gel Extraction Kit)对产物进行胶回收,得到线性化pEn-Chimera入门载体。取上下游靶位点引物TAC1-1-F/R或TAC2-1-F/R各2 μL、ddH2O 46μL于离心管中混匀,95℃处理5 min后取出,室温冷却20 min,制备靶点互补双链。取pEn-Chimera入门载体2μL、互补带有靶点序列的产物3μL、T4连接酶1μL、T4 Buffer 5μL于200μL PCR管中混匀,25℃反应1 h,之后65℃反应15 min,进行载体与靶点序列T4酶的连接反应,最后采用电击法进行DH5α感受态转化,并过夜培养。挑选单菌落,以M13F为上游引物、TAC1-1-R或TAC2-1-R为下游引物,上下游引物各1μL,Premix rTaq 10μL,ddH2O 8 μL,进行序列扩增。扩增程序为:95℃预变性5 min;95℃变性30 s,60℃退火30 s,72℃延伸1 min,35个循环;72℃延伸10 min,4℃保存,1%琼脂糖凝胶电泳进行检测,筛选阳性菌落,测序。
1.3 表达载体的构建
取pEn-Chimera入门载体2μL、pDE-CAS9-NPTⅡ表达载体3μL、TE buffer 4μL、LR clonaseⅡ1μL于PCR管中,25℃反应6 h,构建LR反应。取50μL DH5α感受态细胞,加入LR反应产物,混匀,冰浴30 min,42℃热激45 s,再次冰浴2 min,加入500μL无菌LB培养基,37℃、200 r/min培养1 h,实现感受态细胞的转化。将已转化的感受态细胞在LB固体培养基上均匀涂开,37℃过夜培养。以TAC1-1-F或TAC2-1-F为上游引物、SS102为下游引物,上下游引物各1 μL,Premix rTaq 10μL,ddH2O 8μL,进行PCR扩增。扩增程序为:95℃预变性5 min;95℃变性30 s,60℃退火30 s,72℃延伸1 min,35个循环;72℃延伸10 min,4℃保存,1%琼脂糖凝胶电泳进行检测,阳性菌落进行测序。
1.4 农杆菌的转化
取农杆菌感受态细胞100μL加入0.01~1.00μg表达载体质粒,混匀,冰上静置5 min,液氮冷冻5 min,37℃水浴5 min,冰浴5 min,加入700μL LB液体培养基,28℃培养2~3 h。6 000r/min离心1 min,取100μL上清重悬菌块,在LB固体培养基上均匀涂开,28℃倒置培养2~3 d。取SS61和靶点反向序列TAC1-1-R或TAC2-1-R各1μL,Premix rTaq 10μL,ddH2O 8μL于PCR管中混匀,使用枪头蘸取单菌落于离心管中,PCR扩增程序:95℃预变性5 min;95℃变性30 s,60℃退火30 s,72℃延伸1 min,35个循环;72℃延伸10 min,4℃恒温保存。1%琼脂糖凝胶电泳进行农杆菌转化检测。
1.5 717-1B4杨的转化
利用叶盘法进行717-1B4杨的转化。①预培养:取生长良好、无病害的717-1B4杨组培苗自上而下第三片到第五片叶,使用打孔器沿叶脉方向进行打孔取材,置于CIM愈伤诱导(无抗生素)培养基中28℃暗培养。②菌种活化与培养:取农杆菌于含Rif和Spec的LB培养基上进行划线,28℃倒置培养,筛选单菌落进行摇菌活化,180 r/min、28℃暗培养至OD600为0.3~0.5。③侵染:将培养好的菌液进行离心,重悬,重悬OD600值为0.3~0.5,振荡侵染叶片10 min,50~100 r/min离心,过滤菌体,取出叶片。④共培养:将叶片于CIM愈伤诱导培养基(无抗生素)上,28℃暗培养2~3 d,直至叶片背面出现环形菌斑,但无大面积形成。⑤选择培养:取出共培养的叶片,ddH2O洗涤3次,每次5 min,清洗液洗涤1次,转移至抗Kana的CIM愈伤诱导培养基中,28℃暗培养2周后,更换新的抗Kana的CIM愈伤诱导培养基,转为28℃光下培养1周,促进愈伤的形成。⑥使用Kana抗性的SIM芽诱导培养基进行不定芽诱导,直到不定芽生长至1~2 cm,切下置于Kana抗性SEM芽伸长培养基组培瓶中进行光下伸长培养,直到获得抗性植株。
1.6 转基因植株鉴定
使用CTAB法提取转基因植株幼嫩叶片DNA,以序列TAC1-1-F或TAC2-1-F为上游引物、SS102为下游引物,PCR扩增体系(20 μL):上、下游引物各1μL、DNA 1μL、2×Easy Taq PCR Super Mix 10μL、ddH2O 7μL。PCR扩增程序:95℃预变性3 min;95℃变性30 s,60℃退火30 s,72℃延伸1 min,35个循环;72℃延伸10 min,4℃恒温保存,取10μL PCR产物于1%琼脂糖凝胶电泳进行检测,筛选阳性植株。
1.7 T4载体连接及测序
以转基因植株DNA为模板,使用引物TLZF和TLZ-R进行PCR扩增。扩增体系(20μL):上、下游引物TLZ-F、TLZ-R各1μL,模板DNA 1μL,2×Easy Taq PCR Super Mix 10μL,ddH2O 7μL。扩增程序:95℃预变性3 min;95℃变性30 s,60℃退火30 s,72℃延伸1 min,30个循环;72℃延伸10 min。4℃恒温保存。PCR扩增产物纯化后与pMD19-T载体连接,取5μL连接产物转入大肠杆菌感受态细胞,在AMP抗性的LB培养基上37℃培养12~16 h,挑选完整、圆润的20个单菌落进行菌落检测,取阳性菌斑进行活化、测序,利用DNAMAN(Version 7.0)对测序结果与靶点序列进行比对,计算编辑效率。
2 结果与分析
2.1 TAC基因的靶点设计
利用717-1B4杨基因组数据库及推荐的靶位点,结合已知的水稻、拟南芥基因序列,将717-1B4杨和毛果杨基因组序列进行对比,在毛果杨中确定2个TAC同源基因Potri.002G175300、Potri.014G102600,将其与717-1B4杨中的2个TAC同源基因进行比对,序列相似度分别高达97.65%和98.01%(图1)。表明717-1B4杨TAC基因组序列为TAC-like基因的旁系同源基因。根据基因编辑靶点设计原则,设计了717-1B4杨中2个TAC同源基因的2个单基因编辑靶点,其中第一个同源基因的基因编辑位点位于外显子第323个核苷酸处,第二个同源基因的基因编辑位点位于外显子第58个核苷酸处。靶点均位于基因的前中端。
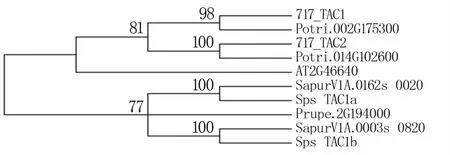
图1 TAC基因家族系统进化树
2.2 TAC基因的入门载体构建
电泳检测结果(图2)表明,目标条带约369 bp,与预期目标片段一致。经测序比对,TAC1-1、TAC2-1基因靶点已成功进入pEn-Chimera载体,表明TAC1-1、TAC2-1入门载体连接成功。
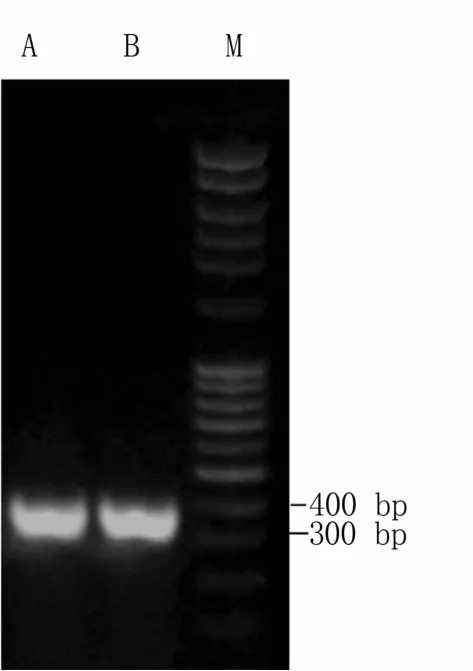
图2 入门载体的PCR鉴定
2.3 TAC基因的表达载体构建
将携带靶点序列的入门载体pEn-Chimera通过同源置换反应构建pDE-CAS9-NPTⅡ表达载体。电泳检测结果(图3)表明,目标条带约933 bp,与预期目标片段一致,且无明显非特异性扩增。经测序分析,TAC1-1、TAC2-1入门载体已成功置换pDE-CAS9-NPTⅡ表达载体中,表明pDE-CAS9-NPTⅡ::TAC1-1、pDE-CAS9-NPTⅡ::TAC2-1表达载体构建成功。
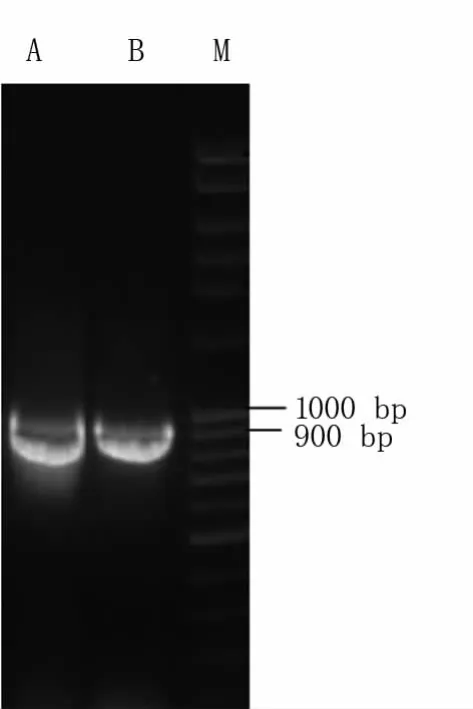
图3 表达载体的PCR鉴定
2.4 转基因植株的获得
将构建成功的基因编辑载体pDE-CAS9-NPTⅡ::TAC1-1、pDE-CAS9-NPTⅡ::TAC2-1通过农杆菌介导法转化717-1B4杨。经外植体叶片预培养、抗性愈伤诱导培养、抗性芽的分化和伸长培养、生根与增殖培养,pDE-CAS9-NPTⅡ::TAC2-1载体成功获得抗性苗(图4)。

图4 转基因植株获得过程
2.5 转基因植株的PCR鉴定
对获得的20株抗性苗进行鉴定,以携带靶点的质粒为阳性对照,野生型717-1B4杨为阴性对照,进行PCR检测。检测结果(图5)表明,1、2、4、6、8、11、12、15、16、18、19、20号共12个株系检测为阳性,PCR阳性鉴定转化率为60%。
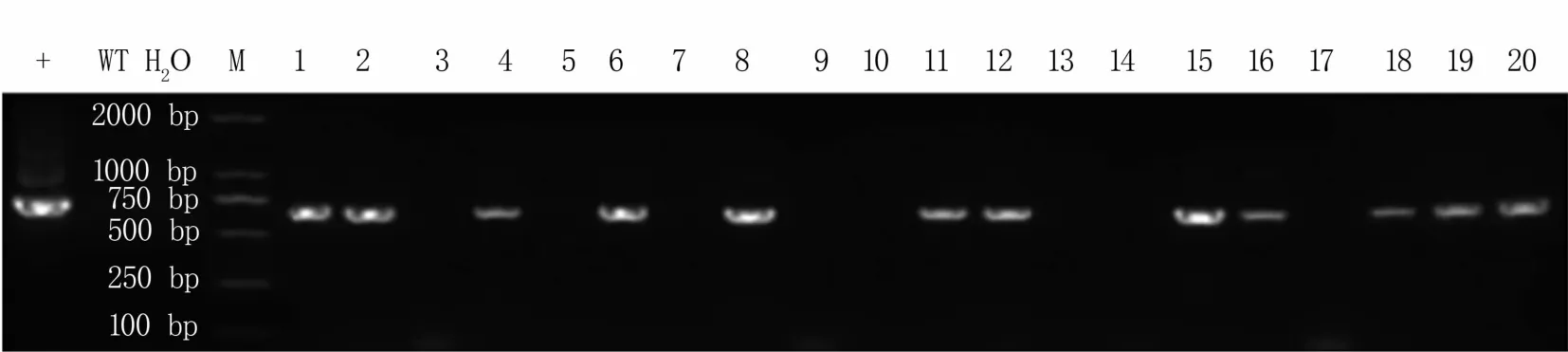
图5 转基因阳性苗鉴定
2.6 转基因植株靶点序列突变分析
在获得的12株转基因植株中,基因靶标序列位点在8号、15号株系中均发生碱基缺失,其中8号株系靶点的第11、12个碱基发生缺失,15号株系靶点的第5~9个碱基发生缺失,靶点编辑效率16.7%,均为杂合突变体。其余株系均如株系1、2、4号未发生基因缺失或插入(图6)。

图6 靶点基因突变类型
3 讨论
TAC基因作为IGT基因家族的一员,主要参与植物地上部的分枝角度以及侧枝生长方向的调控。在水稻、玉米、桃树、拟南芥等植物中,TAC1通过调控枝条的向地性,影响植物的分枝角度[17];在水稻中,TAC2突变体表型与TAC1接近。因此,本试验以TAC基因为例设计了两个基因靶点,构建入门载体和表达载体,通过农杆菌介导法转化717-1B4杨,利用CRISPR/Cas9系统获得抗性植株,并对抗性植株进行鉴定、靶点编辑检测及靶点编辑效率分析,旨在为后续717-1B4杨IGT基因家族TAC基因功能的研究提供数据参考。
CRISPR/Cas9系统作为新兴基因编辑技术,具有广泛的应用前景,仅需设计1~2个目标靶向序列就可以引导Cas9蛋白对目的基因进行特异性的结合修饰,最终导致基因突变或敲除,极大地推动了基因基础研究的发展[18]。研究显示,利用CRISPR/Cas9技术编辑植物基因组DNA,产生的突变类型多为碱基缺失和插入,且碱基缺失突变频率远高于碱基插入突变频率[19,20]。本试验利用CRISPR/Cas9技术进行编辑,从阳性植株中筛选获得2株杂合突变体,且均为碱基缺失,基因编辑效率16.7%,说明717-1B4杨TAC基因产生的碱基缺失突变频率较高,与前人研究结论一致。
本研究构建的两个表达载体为单靶点基因编辑载体,TAC基因的靶位点符合设计要求,GC碱基含量为40%,但靶位点编辑效率较低。有研究指出,通常情况下单个sgRNA只能靶向1个基因位点,编辑效率低且不稳定,而多个sgRNA靶向同一基因的不同位点,不仅可以提高基因编辑频率,还可能造成大片段的碱基突变[21]。胡春华等[22]利用改良的CRISPR/Cas9多靶点载体系统,以香蕉MaPDS基因为例,成功实现了对香蕉内源基因的定点敲除,获得白化突变体株系。汪秉琨等[23]构建了Wx基因双靶点CRISPR/Cas9表达载体,也获得多个位点突变的个体。CRISPR/Cas9系统在植物中的编辑效率受其他多种因素的影响,如Cas9蛋白活性[24]、靶标数量、sgRNA启动子[25]、靶点序列GC含量[26]及载体的sgRNA表达量[27]等,甚至还会出现脱靶情况[28]。Slaymaker等[29]对Cas9蛋白结构进行了完善,提高了表达载体的突变效率,减少了脱靶情况。Ma等[30]在水稻研究中发现靶位点序列的GC含量较高(50%~70%)时编辑效率相对更高。Dang等[31]在原有sgRNA长度的基础上增加了10个碱基,完善了sgRNA骨架序列,结果表明完善后的基因编辑效率提高约50%。Fu等[32]探讨了突变效率与sgRNA长度的关系,发现其自身活性不受影响的前提下,控制sgRNA长度在17~18 nt可以明显降低脱靶效应。以本试验结果为基础,在运用CRISPR/Cas9系统对物种进行编辑时,可以尝试对该物种所用的sgRNA长度、Cas9载体、GC含量、蛋白结构等进行优化,从而获得更高的编辑效率。
本试验pDE-CAS9-NPTⅡ::TAC2-1载体成功获得抗性苗,pDE-CAS9-NPTⅡ::TAC1-1载体未获得抗性苗,植株遗传转化的方法、品种等因素可能是原因之一;717-1B4杨作为试验材料,在遗传转化预培养浸染时发现植株外植体容易发生褐化死亡、农杆菌过度污染导致死亡等问题,对抗性植株的获得造成了极大阻碍,在参考刘闵豪[33]、苏文龙[34]等对预培养时间、菌液侵染浓度以及农杆菌侵染时间等一系列优化调整后,农杆菌过度污染等问题得到缓解。本试验基于CRISPR/Cas9技术,成功构建了717-1B4杨TAC基因敲除表达载体,并获得了基因突变植株,后续将移入土中继续进行观察与分析,为深入研究TAC基因的生物学功能及分子机制提供良好的数据参考。
4 结论
本研究利用717-1B4杨基因组数据构建了TAC基因敲除表达载体,并对717-1B4杨进行了遗传转化,共获得12个株系抗性苗,基因编辑效率为16.7%,为CRISPR/Cas9技术在717-1B4杨中的应用做了初步探索,同时为TAC基因功能的研究和分子育种提供理论依据。
