Newly discovered endocrine functions of the liver
2021-12-06JaneRhyuRunYu
Jane Rhyu,Run Yu
Jane Rhyu,Run Yu,Division of Endocrinology,Diabetes,and Metabolism,UCLA David Geffen School of Medicine,Los Angeles,CA 90095,United States
Abstract The liver,the largest solid visceral organ of the body,has numerous endocrine functions,such as direct hormone and hepatokine production,hormone metabolism,synthesis of binding proteins,and processing and redistribution of metabolic fuels.In the last 10 years,many new endocrine functions of the liver have been discovered.Advances in the classical endocrine functions include delineation of mechanisms of liver production of endocrine hormones[including 25-hydroxyvitamin D,insulin-like growth factor 1(IGF-1),and angiotensinogen],hepatic metabolism of hormones(including thyroid hormones,glucagon-like peptide-1,and steroid hormones),and actions of specific binding proteins to glucocorticoids,sex steroids,and thyroid hormones.These studies have furthered insight into cirrhosis-associated endocrinopathies,such as hypogonadism,osteoporosis,IGF-1 deficiency,vitamin D deficiency,alterations in glucose and lipid homeostasis,and controversially relative adrenal insufficiency.Several novel endocrine functions of the liver have also been unraveled,elucidating the liver’s key negative feedback regulatory role in the pancreatic α cell-liver axis,which regulates pancreatic α cell mass,glucagon secretion,and circulating amino acid levels.Betatrophin and other hepatokines,such as fetuin-A and fibroblast growth factor 21,have also been discovered to play important endocrine roles in modulating insulin sensitivity,lipid metabolism,and body weight.It is expected that more endocrine functions of the liver will be revealed in the near future.
Key Words:Liver;Endocrine function;Hormone;Amino acids;Hepatokine;Fibroblast growth factor 21
INTRODUCTION
The liver is a dynamic endocrine organ and mediates critical metabolic pathwaysviaroles in direct hormone and hepatokine production,hormone metabolism,synthesis of binding proteins,detoxification,and processing and redistribution of metabolic fuels[1-4].It participates in multiple signaling pathways with other endocrine organs,including the pituitary,pancreas,gut,thyroid,adrenal glands,and bone,with hormones in turn modulating the liver’s metabolic and synthetic functions[1,5].Diseases that affect the liver lead to a variety of endocrine manifestations,including hypogonadism,osteoporosis,effects on glucose metabolism and growth hormone(GH),and controversial effects on cortisol[1,5].
The liver,with its vascularity,is well-positioned to provide and receive endocrine signals,including those from pancreatic and gut hormones[6].It also receives exposure to antigen-rich blood systemically and from the gastrointestinal system as a lymphoid organ[7]and serves as a principal organ in drug metabolism and clearance[8].Despite only representing 2.5% of the body weight,the liver receives up to 25% of the total cardiac output at rest[9].It also receives a unique double afferent blood flow from the hepatic artery and partially deoxygenated portal vein,with around 75% of the blood flow from the latter[9].The portal vein,in turn,receives blood from the stomach,small and large intestines,pancreas,spleen,and gallbladder[9],with direct physiological implications on the regulation of metabolism by endocrine liver functions[6].Great progress has been made in the understanding of the endocrine functions of the liver in the last 10 years.
ADVANCES IN CLASSIC ENDOCRINE FUNCTIONS OF THE LIVER
We will first briefly summarize the advances in the understanding of the liver classic endocrine functions(Table 1).

Table 1 Classic endocrine functions of the liver
Direct hormone production
The liver directly synthesizes multiple hormones,including 25-hydroxyvitamin D,insulin-like growth factor 1(IGF-1),and angiotensinogen.Given roles in direct hormone production,the liver also has permissive roles of normal hormone function,in particular with effects on bone health,the GH-IGF-1 axis,and renin-angiotensinaldosterone(RAA)pathway.
Vitamin D:The liver is the primary site of 25-hydroxylation of vitamin D to 25-hydroxyvitamin D(calcidiol),the main storage form of vitamin D[10].Vitamin D is a secosteroid hormone well known for its role in calcium and bone homeostasis,with pleiotropic effects on cellular proliferation,differentiation,and immunomodulation[11-13].25-hydroxyvitamin D(calcidiol)then undergoes 1-alpha-hydroxylation in the kidney to the activated form 1,25-dihydroxyvitamin D(calcitriol)[10],which provides the active hormonal effects of vitamin D.The hydroxylation of vitamin D to produce calcidiol is mainly carried out in the liver by multiple cytochrome P450 mixed-function oxidases(CYPs)located in the mitochondria,endoplasmic reticulum(ER),and microsomes,though studies also show presence of these CYPs in extrahepatic tissues[10,11].
IGF-1:The liver is the primary source of IGF-1,a 70-amino acid polypeptide hormone with endocrine,paracrine,and autocrine effects[14].IGF-1 affects almost every tissue and organ[15],and its receptors are ubiquitously expressed[16].Besides mediating the actions of GH,more recently,non-growth-related actions of IGF-1 are found.IGF-1 binds to the insulin receptor and the hybrid IGF-1/insulin receptors,with implications on the metabolic effects of IGF-1[14].IGF-1,GH,and insulin are hypothesized to constitute a regulated axis to inform cells about nutritional status,helping direct cells grow and differentiatevsinduce a state of quiescence,senescence or apoptosis[14].The IGF-1 receptor also participates in a crosstalk with the thyrotropin receptor by forming heterodimers[17],with implications on cellular growth and pathological implications in Graves’ eye disease.
Angiotensinogen:The liver is the primary source of angiotensinogen,which is involved in the RAA system[18].The RAA system is vital for maintaining blood pressure homeostasis,viaeffects on sodium balance,intra- and extra-vascular volume,and systemic vascular tone[19].Angiotensinogen,an alpha-globulin,is the only known substrate for renin and the main precursor molecule for angiotensin II(AngII),the major biologically active peptide in the RAA pathway[19].Despite local tissue production of AngII,liver angiotensinogen is the primary source of renal AngII[18].Hepatocytes tonically secrete angiotensinogen and primarily determine plasma angiotensinogen levels,with small increases in angiotensinogen levels increasing blood pressure and AngII levels[20].
Hormone metabolism
The liver is involved in the metabolism of multiple endocrine hormones,including thyroid hormones,glucagon-like peptide-1,and steroid hormones,with roles in both activation and inactivation of the hormones.
Thyroid hormone:Hepatic metabolism has roles in both activation and inactivation of thyroid hormones.The biologic activity of thyroid hormone is mainly mediated through the active thyroid hormone T3.The thyroid only secretes 20% of the daily T3 requirement,with the remainder 80% converted from T4 by peripheral seleniumcontaining deiodinase enzymes(DIO),of which three primary deiodinases(type 1,2,and 3)have been identified[21].The liver expresses DIO1,along with the kidney and thyroid,which converts T4 to T3,though with less kinetic efficiency compared to DIO2,which is expressed by brown adipose tissue and the pituitary.Subsequently,the thyroid hormone is metabolized by conjugation with sulfate or glucuronic acid,which occurs prominently in the liver[22].
Glucagon-like peptide 1:With the discovery of glucagon-like peptide 1(GLP-1),increasing research has been studying the gut-pancreas-liver axis,and the liver has been shown to play a key role in the hormone’s metabolism[23].GLP-1 is an incretin hormone produced by the intestinal L-cells in response to ingestion of nutrients,including carbohydrates,fatty acids,and fiber[24].It stimulates insulin secretion in a glucose-dependent manner,with associated inhibition of hepatic gluconeogenesis,and promotes insulin gene transcription and growth and proliferation of islet cells[24].GLP-1 is inactivated by dipeptidyl peptidase-4(DPPIV),also known as CD26,a ubiquitous membrane-associated peptidase[25].DPPIV has pleiotropic effects and widespread tissue distribution in all organs,with expression in capillary endothelial cells and high expression in the liver[25].
Steroid hormone metabolism:The liver participates in most steps of steroid hormone regulation,starting from being the primary site of cholesterol biosynthesis[26,27].At the liver,steroid hormones undergo phase I metabolism by cytochrome P450 enzymes(CYPs),viamultiple pathways including hydroxylation or reduction,and phase II metabolism,alsoviavarious processes including glucuronidation,sulfation,or methylation[27],ultimately leading to excretion of their conjugates in urine or bile.
Steroid hormone metabolism:Sex hormones:The liver is the main site for metabolic conversion of estrogens,progesterone,and androgens to their metabolitesviaCYPs,which are abundantly expressed in the liver[28].In particular,as part of the first phase of metabolism,estrogens undergo hydroxylation by numerous CYPs,including 2-hydroxylation to 2-hydroxyestradiol and 4-hydroxylation to 4-hydroxestradiol,which represent 80% and 20% of biotransformation of estradiol in the liver,respectively.2-hydroxylation is mainly catalyzed by CYP1A2 and CYP3A4,which are expressed in the liver,and CYP1A1 in extrahepatic tissues[28].4-hydroxestradiol,unlike 2-hydroxestradiol,is associated with free radical generation and cellular damage,with associated increased risk of carcinogenesis in the breast and endometrium.Subsequent phase II metabolism of sex hormones,viaO-methylation by catechol O-methyltransferase(COMT),glucuronidation,or sulfation,occurs at high levels at the liver,with subsequent elimination in the urine or stool[28-30].
Steroid hormone metabolism:Glucocorticoids and mineralocorticoids:The liver is also the primary site of glucocorticoid and mineralocorticoid metabolism[27].Cortisol is converted to and from its inactive metabolite cortisone by two isozymes of 11-beta hydroxysteroid dehydrogenase(11-beta-HSD)[31].11-beta-HSD type 1(11-beta-HSD1)is widely distributed,though most abundantly located in the liver and adipose tissue,and is responsible for converting cortisone back to cortisol[31],within vitroactivity being greater in omental than subcutaneous adipose tissue[32].In healthy individuals,local splanchnic cortisol production,including from the liver,can equal or even exceed that produced by extra-splanchnic tissues,including the adrenal gland[32].In obese,non-diabetic individuals,the liver has been shown to account for virtually all splanchnic cortisol production[32].Though primarily secreted from the adrenal glands under the regulation of the RAA axis,animal studies suggest possibility of local hepatic aldosterone production during liver injury,which may contribute to fibrogenesis[33].Glucocorticoids and mineralocorticoids,like other steroid hormones,undergo phase I and phase II metabolism in the liver,with excretion of their conjugates in urine or bile[27].
Binding protein production
Lipophilic hormones,including steroid hormones,are not water soluble and need to be carried in the blood stream by binding proteins[2,34].The liver is the primary source of binding proteins for many hormones.The liver produces specific binding proteins to multiple lipophilic hormones,including glucocorticoids,mineralocorticoids,sex steroids,thyroid hormones(T3 and T4),and vitamin D metabolites[2,34].Binding globulins for these lipophilic hormones include cortisol binding globulin(CBG,which binds cortisol,aldosterone,and progesterone),sex hormone binding globulin(SHBG,which binds estradiol,testosterone,and other sex hormones),thyroxine binding globulin(TBG,which binds T3 and T4),and vitamin D binding globulin(DBG,which binds vitamin D metabolites)[2,34].Binding proteins that are produced by the liver also include transthyretin(which binds thyroid hormone and retinol),IGF-1 binding proteins(IGFBP,which binds IGF,including IGF-1),and nonspecific binding proteins including albumin and lipoproteins.Binding proteins serve as a circulating reservoir for hormones,potentially regulating tissue distribution and target destination in a manner that can be highly selective and targeted[2,35].Binding protein expression and production,which occur primarily at the liver,is complex and under the regulation and influence of multiple factors[2].Most binding protein expression increase in response to estrogens,including physiologically with pregnancy or with oral contraceptives[2,34].Hepatic failure and protein-losing nephropathies lead to decrease of binding proteins in general[2,34].
Endocrine dysregulation in liver disease
The liver mediates the effects of numerous hormonal pathways,whether directly or indirectly;thus,not surprisingly,derangements affecting the liver lead to disruptions of various hormonal pathways.Patients with cirrhosis are characterized by various endocrinopathies,including relative increase in estrogen compared to androgens,hypogonadism,osteoporosis,IGF-1 deficiency,vitamin D deficiency,alterations in glucose and lipid homeostasis,and perhaps more controversially a relative adrenal insufficiency.
Sex hormones:Cirrhosis is characterized by symptoms of estrogen-androgen imbalance,with relatively higher estradiol and lower testosterone concentrations[36].The etiology of estrogen-testosterone imbalance is at least in part due to conversion of androgens to estrogens in cirrhosis,which in large part occurs peripherally[36].The pathophysiology of hypogonadism is complex,including potential contribution from hypothalamic-pituitary suppression from a relatively increased estrogen circulation.SHBG is elevated in compensated cirrhotic patients,with subsequent decreases with decompensated cirrhosis,leading to concern for potential underestimation of hypogonadism in cirrhosis[34].
Cortisol:Patients with cirrhosis have relatively lower cortisol levels,also in the setting of lower production of cortisol binding globulin[37].Some studies suggest the presence of a relative adrenal insufficiency in cirrhosis,also termed critical illnessassociated corticosteroid insufficiency[38].These studies suggest a potential hepatoadrenal syndrome in advanced liver disease,with associated inadequate cortisol production during stress response[38].The decrease in cortisol binding globulin makes the diagnosis more difficult,though some studies suggest that free cortisol levels are decreased in relative adrenal insufficiency[37].Hepatoadrenal syndrome and associated low free cortisol are attributed to decreased formation of HDL precursors and formation of proinflammatory cytokines and endotoxins[38].
RAA system:In liver disease,the systemic RAA pathway is upregulated due to systemic and splanchnic arterial vasodilation and associated hypoperfusion of the renal system[39].Notably,the cirrhotic liver is able to produce angiotensinogen to near-normal plasma levels until the end stages[40].
DPPIV and GLP-1:DPPIV may play a role in linking type 2 diabetes with chronic liver disease.Type 2 diabetes has been associated with a greater than 2-fold increased risk of liver disease[41],andin vitrostudies have suggested that elevated glucose can induce DPPIV expression in liver cells[42].The increased DPPIV activity,which degrades the incretin hormone GLP-1,may contribute towards development of IGT,insulin resistance,lipogenesis,and hepatic injury in liver disease[25,43].Serum DPPIV levels are notably increased in cirrhosis[25],and increased DPPIV expression in the liver has been observed in hepatitis C,NAFLD,experimental liver regeneration,and cirrhosis[25,43].Cirrhotic nodules show diffuse and uniform staining of DPPIV,with loss of usual zonal expression of DPPIV[43],and degree of hepatic expression of DPPIV has also been shown to correlate with NAFLD grading[25].Increased DPPIV expression has also been seen in various malignant tumors,including hepatocellular carcinoma,with DPPIV noted to promote resistance to anticancer agents[25].
Thyroid hormone:Given the liver’s role in thyroid hormone metabolism,including local conversion of T4 to T3 by DIO1[21],patients with cirrhosis may present with abnormalities in thyroid hormone levels[44].Though a variety of patterns are seen,the most common pattern is a low total T3(TT3),low free T3(FT3),elevated reverse T3(rT3),low total T4(TT4),variable literature on elevatedvslow free T4(FT4)levels,and possible elevations in TSH[44,45].The low total hormone levels are attributable to low TBG[44].The pattern is consistent with low T3 syndrome,which occurs in systemic illnesses,and represents non-thyroidal illness syndrome,previously known as euthyroid sick syndrome[44].
IGF-1:Systemic IGF-1 deficiency in cirrhosis has been associated with an altered metabolic profile,including diabetes,deregulated lipid profile,and cardiovascular disease[14].Lack of liver-derived IGF-1,in particular,has been associated with resultant insulin insensitivity in the liver,skeletal muscle,and adipose tissue,and corresponding hyperinsulinemia[46].In NAFLD,the severity of steatosis has been correlated with a decrease in IGF-1 levels,with statistically significant differences in IGF-1 levels between mild-moderatevssevere steatosis[14,47].
Bone health and vitamin D:Chronic liver disease,including cirrhosis regardless of etiology,is associated with osteomalacia,osteopenia,and osteoporosis,and up to 40% of patients with chronic liver disease may develop an osteoporotic fracture[48].The etiology of hepatic osteodystrophy is not well understood,though potential contributing factors include hypogonadism,and decreased hepatic production of IGF-1 and fibronectin[48].There is a shift in cytokine production with changes in the receptor activator of nuclear factor kappa-B ligand(RANKL)/osteoprotegerin(OPG)system and an up-regulation of IL-6,which stimulates osteoclasts[48].Decreased vitamin D synthesis,which is more marked in severely compromised liver function or in cholestatic liver disease,can further contribute to increased osteoporotic risk[49].History of steroid treatment in chronic liver disease may be a risk factor for osteoporosis as well[48,49].Different etiologies of liver disease may differ in their pathogenesis of osteoporosis,and in particular,diseases such as hemochromatosis and Wilson’s may also directly impact bone health[48].
NOVEL ENDOCRINE FUNCTIONS OF THE LIVER
Besides the advances in the understanding of classic endocrine functions of the liver,novel liver endocrine functions have been unraveled in the last several years(Table 2),including endocrine regulation of pancreatic α cells,adipose tissue,and insulin sensitivity.

Table 2 Novel endocrine functions of the liver
Feedback regulation of pancreatic α cells and glucagon
A major novel endocrine function of the liver is its critical role in a pancreatic α cellliver axis that regulates pancreatic α cell proliferation and circulating glucagon and amino acid levels[50,51].The pancreatic α cells,unlike the insulin-secreting β cells,have been considered a mysterious cell type until recently[52,53].The α cells appear first during embryogenesis[54].The main known function of the α cells is to produce and secrete the hormone glucagon[55].Glucagon raises circulating glucose levels directly by stimulating gluconeogenesis and glycogenolysis,and indirectly by inhibiting insulin secretion[55,56].
Recently,a new α cell-liver axis has been discovered,endowing the liver with new endocrine functions[50,51].The first clue of the α cell-liver axis came from glucagon receptor(GCGR)knockout mice[57,58].The GCGR knockout mice harbor diffusely enlarged pancreas and exhibit extremely high glucagon levels[57-59].Histologically,the pancreas of GCGR knockout mice contain numerous islets at various sizes,which are composed of mostly α cells as demonstrated by immunochemistry[57-59].Normally the number of islets is quite small,and the islets are mostly composed of β cells.Mahvash disease,a human autosomal recessive hereditary disease discovered by our group,is caused by biallelic inactivating GCGR mutations,and its universal features are also α cell hyperplasia and hyperglucagonemia[60-62].GCGR inactivation in zebra fish and non-human primates also result in α cell hyperplasia and hyperglucagonemia[63-66].Thus,preservation of glucagon function is conserved throughout evolution.
Although a physiological compensation of hyperglucagonemia in animals and humans with inactive GCGR is quite intuitive,the specific mechanism of the compensation was initially not clear[67].The liver-specific GCGR knockout mice interestingly have similar α cell hyperplasia and hyperglucagonemia,as those in global GCGR knockout mice[57,58,68],suggesting that the liver is the only target organ of glucagon that sends feedback signals to α cells,and that loss of the usual negative feedback mechanism stimulates α cell hyperplasia and glucagon secretion.This theory is also supported by the liver-specific stimulatory G protein α subunit(Gsα)knockout mice,which also exhibit α cell hyperplasia and hyperglucagonemia[69].As glucagon antagonists were a promising anti-diabetes medication,both academia and pharmaco-logical companies became interested in the α cell-liver axis due to potential applications in diabetes drug development[70,71].Some of the key original large-scale experiments leading to the discovery of the role of amino acids in regulating α cells were performed by pharmaceutical companies[72-74].
The liver may regulate α cellsvianeural or humoral mechanisms[67,68].Islet transplantation experiments demonstrate that the liver uses a humoral mechanism[68].Wild-type islets transplanted into the kidney of GCGR knockout mice undergo α cell hyperplasia,while GCGR knockout islets transplanted into wild-type kidney undergo reduced α cell proliferation.Thus,it is assumed that the liver sends a humoral factor(hormone)to stimulate pancreatic α cells,a phenomenon that is pronounced in diseases where the usual negative feedback mechanism is affected.
Initially,it was hoped that a single liver hormone would be isolated from differential liver gene expression patterns of wild-type and GCGR knockout mice[67].Several groups,including ours,performed liver mRNA arrays of GCGR knockout mice and in wild-type mice treated with inhibitory GCGR antibodies,using wild-type mice as control[67,68,72].Not surprisingly,many genes are overexpressed(potential stimulatory hormones)or underexpressed(potential inhibitory hormones)in the GCGR knockout liver[67,68,72].Genes involved in gluconeogenesis are downregulated in the GCGR knockout liver[67,68,72].On the other hand,genes involved in amino acid synthesis(e.g.,asparagine synthetase,Asns)are upregulated,and genes involved in amino acid catabolism(e.g.,glutaminase 2,Gls2)are downregulated[67,68,72].Genes regulating lipid metabolism are also differentially expressed[67,68,72].Most of the genes with significant differential expression were not bona fide hormone candidates because they were not secreted proteins[67,68,72].InhbA and DefB1 were the only 2 overexpressed secreted proteins by both the GCGR knockout liver and wildtype liver treated with inhibitory GCGR antibodies;however,these two proteins were are also upregulated by glucagon in primary hepatocytes and thus unlikely the pursued liver hormone[67,68,75].
Another possibility was that the liver hormone may not be a direct gene product such as a protein or polypeptide;rather,the hormone may be a small molecule or metabolite[67].Metabolomes of the GCGR knockout and wild-type mice were compared[72].Many differences exist but most notable differences were in glucose,amino acid,nucleotide,and bile acid levels[72].The GCGR knockout mice have lower glucose levels(70% of wild-type value)and higher levels of most amino acids(up to 15-fold for alanine,glutamine,glycine,lysine,and threonine)and 2 bile acids(cholic acid and glycocholic acid,both about 200-fold)[72].In humans with Mahvash disease,glucose levels are generally normal,but the levels of amino acids,especially alanine and glutamine,are clearly elevated[62,76-78].
Pinpointing the identity of the novel liver hormone requires tremendous amount of work.Parabiosis of GCGR knockout and wild-type mice was considered,but no such models were published[67].A more practicalin vitroislet culture assay was adopted by most groups to screen for the liver hormone that stimulates α cell hyperplasia and hyperglucagonemia[73-75].With the islet culture assay,it is shown that a < 10 kDa fraction of serum from GCGR knockout mice sufficiently stimulates α cell proliferation[75].This fraction contains small proteins or peptides,lipids,amino acids,and metabolites[75].We have discussed earlier that most proteins or peptides are unlikely the liver hormone.Eliminating lipids from the fraction does not change the activity of the fraction in stimulating α cell proliferation[75].Finally,as amino acids levels are much higher in GCGR knockout serum,cocktails that mimic the amino acids levels in GCGR knockout mice serum have been tested for their ability to stimulate α cell proliferation,and indeed they do[73-75].
Individual amino acids were further tested to see if a particular amino acid is sufficient to stimulate α cell proliferation[73-75,79].So far,the data on individual amino acids are still somewhat controversial.Most individual amino acid do not stimulate α cell proliferation or glucagon secretion[73-75,79].Glutamine alone stimulated α cell proliferation in 2 studies,but it did not stimulate glucagon secretion in another,which is intriguing as α cell hyperplasia and hyperglucagonemia coexist in all models of GCGR inhibition[74,75,79].Alanine alone stimulated α cell proliferation in one study,but not in another,albeit acutely stimulating glucagon release[75,79].Experimental conditions may explain some of the different results.It is also possible that α cell proliferation and acute glucagon release may be separate processes.
The α cell receptor for amino acids is under active research.In GCCR knockout mice and in wild-type type mice treated with inhibitory GCGR antibodies,the most upregulated α cell gene is the amino acid transporter Slc38a5(20-80-fold increase)[74,75].Slc38a5 preferentially transports glutamine and several other amino acids,which is concordant with the stimulatory effect of glutamine on α cell proliferation[74,75].Slc38a5 knockout mice treated with inhibitory glucagon antibodies and Slc38a5 and GCGR double knockout mice exhibited less prominent α cell hyperplasia( approximately 50% less)but similar hyperglucagonemia[74];this data suggested that Slc38a5 is at least partially responsible for amino acid-stimulated α cell hyperplasia and that α cell hyperplasia and hyperglucagonemia may be regulated separately.Slc38a5,however,is not expressed in human α cells[74].Another amino acid transporter Slc38a4 is enriched in human α cells when mice with human islet implants are treated with inhibitory GCGR antibodies[80].In humans with Mahvash disease,Slc38a4 is expressed in the α cells[80],supporting a role of the amino acid transporter in mediating amino acid-stimulated α cell hyperplasia in humans as well.The mTOR pathway in α cells is activated by amino acids as well,contributing to α cell hyperplasia[73-75].
As a result of these studies,the α cell-liver axis has largely been clarified(Figure 1).The α cells secrete glucagon,which signals the liver to increase hepatic amino acid breakdown and reduce amino acid synthesis,consequently leading to desirable amino acid levels in the circulation.After glucagon signaling is inhibited,the liver decreases amino acid breakdown and increases amino acid synthesis,thus raising circulating amino acid levels.The amino acid levels,in turn,act on the α cell amino acid transporters to stimulate α cell proliferation.The evolutionarily conserved α cell-liver axis suggests that glucagon’s primary role may be regulating amino acid levels.
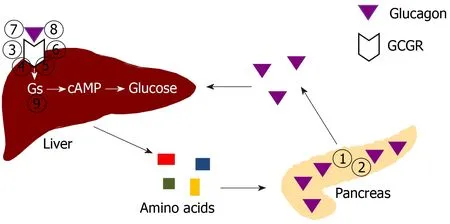
Figure 1 Schematic drawing of regulation of pancreatic α cell number and glucagon secretion by amino acid levels controlled by the liver.The numbers indicate specific ways to disrupt glucagon signaling.(1)Glucagon deletion;(2)Prohormone convertase 2 deletion(with no mature glucagon secretion);(3)Glucagon receptor(GCGR)global deletion;(4)GCGR liver-specific deletion;(5)GCGR inactivating mutation;(6)GCGR antisense RNA;(7)GCGR antagonists;(8)GCGR antibodies;and(9)Gsα liver-specific deletion.See text for details.Citation:Yu R,Zheng Y,Lucas MB,Tong YG.Elusive liver factor that causes pancreatic α cell hyperplasia:A review of literature.World J Gastrointest Pathophysiol 2015;6(4):131-139.Copyright ©The Author(s)2015.Published by Baishideng Publishing Group Inc[67].GCGR:Glucagon receptor.
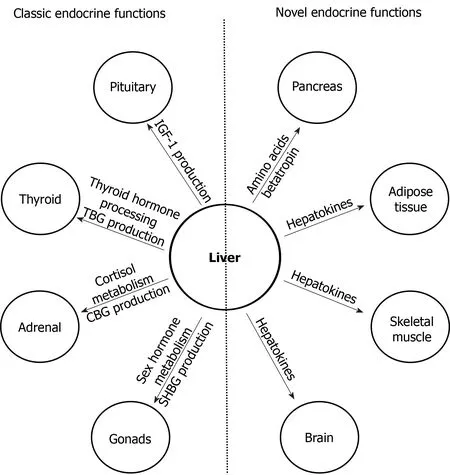
Figure 2 Major classic and novel endocrine functions of the liver.Left,major classic endocrine functions of the liver;right,novel endocrine functions of the liver.See text for details.IGF-1:Insulin-like growth factor 1;TBG:Thyroxine binding globulin;CBG:Cortisol binding globulin;SHBG:Sex hormone binding globulin.
Betatrophin
Betatrophin(also known as angiopoietin-like protein 8,ANGPTL8)is a 22-kD protein produced and secreted by the liver and adipose tissue[81,82].Several years ago,betatrophin was touted as the long sought-after liver hormone that stimulates pancreatic β cell proliferation and insulin production in conditions with insulin resistance[83,84].An insulin resistance mouse model based on insulin receptor antagonist(S961)infusion exhibits remarkable hyperinsulinemia and beta cell hyperproliferation[83].As S961 does not directly stimulate β cell proliferation,it was hypothesized that a humoral factor mediates the stimulation of β cell proliferation in this mouse model[83].Screening of liver genes that were differentially expressed as a result of S961 infusion suggested that betatrophin,a secreted protein that is upregulated by S961 infusion,could be the humoral factor[83].Betatrophin expression correlated well with β cell proliferation rates.The original report found that liver overexpression of betatrophin stimulated β cell proliferation[83].
The potential of betatrophin as the Holy Grail for diabetes treatment attracted much attention,but later experiments strongly argue against this function of betatrophin[85-87].Betatrophin knockout mice exhibited normal glucose metabolism and similar hyperinsulinemia and β cell hyperproliferation in response to S961 infusion[85,86].Detailed analysis of pancreas morphometry by several laboratories definitively showed that betatrophin overexpression does not stimulate β cell proliferation[88].The only exception was that direct delivery of betatrophin to pancreas does stimulate β cell proliferation in rats[89].In some mouse models of diabetes,betatrophin lowered glucose levels without effects on β cell proliferation[90].Overall,betatrophin,despite the name,does not appear to stimulate β cell proliferation.
Betatrophin,however,could be a circulating marker of insulin resistance[82].Early studies of betatrophin levels in various forms of human insulin resistance were quite conflictory,partly due to the differences in measurement methods[82].Later studies using more standardized methods for measuring betatrophin were summarized by several meta-analyses on the correlation of circulating betatrophin levels and type 2 diabetes,gestational diabetes,polycystic ovary syndrome(PCOS),and obesity—all conditions with insulin resistance[91-95].Xuet al[91]analyzed 25 such studies and showed a positive and significant correlation between circulating betatrophin levels and insulin resistance.Yueet al[92]analyzed 11 studies on betatrophin in type 2 diabetes and found that betatrophin is significantly elevated in type 2 diabetes.Konget al[93]analyzed 8 studies on betatrophin in gestational diabetes and concluded that betatrophin is significantly elevated in gestational diabetes.Varikasuvuet al[94]analyzed 11 studies on betatrophin in PCOS and concluded that betatrophin is significantly elevated in PCOS.Similarly,Yeet al[95]analyzed 6 studies on betatrophin in obesity and concluded that betatrophin is significantly elevated in obesity.Thus,overall,circulating betatrophin is likely a marker of insulin resistance in humans.The high betatrophin liver expression in mice treated with S961,in retrospect,could simply be a sign of insulin resistance caused by S961[83].It is,however,not clear how insulin resistance upregulates betatrophin.In humans,hyperinsulinemia,often associated with insulin resistance,and metformin,an insulin sensitizer,both decrease betatrophin levels,suggesting that insulin resistance per se upregulates betatrophin levels[96].Betatrophin overexpression could further worsen hepatocyte sensitivity to insulin,the significance of which needs to be further explored[97].
Betatrophin also has a role in lipids regulation[98].Betatrophin knockout mice exhibit much reduced triglyceride levels due to reduction in liver VLDL secretion[86];betatrophin also forms a complex with ANGPTL3,which inhibits lipoprotein lipase(LPL)activity[86].The increased production of VLDL and decreased LPL activity both contribute to hypertriglyceridemia.Betatrophin overexpression doubles triglyceride levels in mice[86].In humans,circulating betatrophin levels are positively correlated with triglyceride levels in the general population[99].In people with dyslipidemia,however,betatrophin levels were lower than in controls[100].Betatrophin may potentially be a target in dyslipidemia treatment[101].
Hepatokines
Hepatokines are metabolism-regulating proteins produced and secreted by the liver[102,103].Several hepatokines have been reported and studied.Five of the most studied hepatokines are discussed in this review:Fetuin-A,fibroblast growth factor 21(FGF21),activin E,Tsukushi,and glycoprotein nonmetastatic melanoma protein B(GPNMB).
Fetuin-A:Fetuin-A,also known as α2-Heremans-Schmid glycoprotein in humans,is one of the first discovered hepatokines[104].A 52-kD glycoprotein,fetuin-A has diverse metabolic functions[104].Under physiological conditions,fetuin-A mostly functions as a carrier protein and regulates osteogenesis and inhibits extra-skeletal calcification[105].Fetuin-A’s role in regulating insulin sensitivity has also been studied in detail[106,107].Fetuin-A knockout mice exhibit higher insulin sensitivity and have less tendency to develop obesity[106].At the molecular level,fetuin-A inhibits insulin receptor phosphorylation in myocytes and adipocytes and adiponectin expression in adipocytes[107].Fetuin-A levels are elevated in patients with insulin resistance or type 2 diabetes,likely mediated by high free fatty acid levels,and high fetuin-A levels are a risk factor for type 2 diabetes[108,109].The thiazolidinedione-type diabetes medication pioglitazone directly inhibits hepatic production of fetuin-A,partly contributing to its action in improving insulin sensitivity[110].
FGF21:FGF21 is a hepatokine that was first discovered in 2000,but its metabolic regulation functions were not characterized until recently[111,112].Although FGF21 is also expressed in adipose tissue and the pancreas,circulating FGF21 is predominantly derived from the liver[113].Hepatic FGF21 expression is regulated by a number of physiological conditions and factors[114].Prolonged starvation(> 7 d)and overnutrition both upregulate FGF21 expression[115,116].Glucagon and the thyroid hormone triiodothyronine(T3)both stimulate FGF21 expression,while insulin may inhibit FGF21 expression in liver[117,118].High-carbohydrate,high-fat diet,and low protein diets stimulate FGF21 expression as well[119,120].The microRNAs miR-577 and miR-212 target FGF21 mRNA for degradation,thus suppressing FGF21 expression[121,122].FGF21 is also upregulated by ER stress[123].At the molecular level,at least some of the above actions are mediated by the nuclear hormone receptor peroxisome proliferation-activated receptor α(PPARα),which binds to regions of the FGF21 promoter and simulates FGF21 expression[124-126].
The human pre-FGF21(precursor of mature FGF21)includes a 28-amino-acid signaling peptide and a 181-amino-acid FGF21 proper as the circulating form[127].FGF21 signals through its transmembrane tyrosine kinase receptors,FGFR1c and FGFR3c,and its transmembrane co-receptor,Klotho-β(KLB)[128].FGF21 downstream signaling is tissue-specific but generally leads to metabolic benefits such as increased insulin sensitivity and weight loss[129].In the adipose tissue,FGF21 stimulates the Ras/Raf/MAPK pathway,with phosphorylation of ERK1 and ERK2,and the mTOR pathway,contributing to higher insulin sensitivity[130-132].Other FGF21 metabolic benefits such as weight loss is mediated by non-adipose tissue such as the brain[133].FGF21 has been a major interest of metabolic drug development.As the native FGF21 is not stable in the usual formulation,re-engineered FGF21 analogues and PEGylated FGF21 have been developed to be more stable[134].Activating monoclonal antibodies targeting FGFR1-β-klotho have also been developed[135].Preclinical and clinical studies have demonstrated clear metabolic benefits of the FGF21 analogs and activating antibodies,such as appetite suppression,weight loss,improved glycemia,and favorable lipid profile[134,135].
Activin E:Activin E belongs to the family of transforming growth factor-β(TGF-β)proteins[136].Activin E is a secreted homodimer of inhibin-βE,which is mainly expressed in the liver[137].Each mature inhibin-βE monomer has 113 amino acids[137].In both mice and humans,inhibin-βE is upregulated by obesity and insulin resistance[138].In mice,hepatic overexpression of inhibin-βE prevents excess weight gain and improves insulin sensitivity by promoting energy expenditureviaincreased fat oxidation[139,140].Inhibin-βE ablation in mice gives conflictory results[138,139].In one study using the transcriptional activator-like effector nucleases(TALENs)to remove liver specific inhibin-βE expression,inhibin-βE-deficient mice exhibited normal weight but had impaired thermogenesis during cold exposure[139].In another study,however,use of small interfering RNA(siRNA)to silence Inhibin-βE expression in the liver reduced weight gain in obese mice[138].Thus,the roles of Activin E in metabolic regulation are still controversial.
Tsukushi:Tsukushi belongs to the family of small leucine-rich proteoglycan(SLRP)extracellular matrix proteins[141].The secreted human Tsukushi protein has 337 amino acids.Besides its role in regulating embryonic development,Tsukushi is found to be a hepatokine,potentially regulating adipose tissue,weight,and energy expenditure[142].In both mice and humans,Tsukushi is upregulated by thyroid hormone[142,143];in mice,Tsukushi is induced by obesity and cold exposure[142].Tsukushi deficiency in mice protects them from diet-induced obesity by increasing adipose tissue thermogenesis and energy expenditure[142].Using mice from a different genetic background,another group could not reproduce the metabolic benefits of Tsukushi deficiency[144].Furthermore,studies have also failed to show deleterious metabolic effects from Tsukushi overexpression[144].The roles of Tsukushi in metabolic regulation thus also remain controversial.
GPNMB:GPNMB is a transmembrane glycoprotein expressed in the liver and other organs[145].The cleaved extracellular domain of GPNMB(a glycosylated 480-aminoacid protein)is a hepatokine targeting adipose tissue[146,147].In 2 obese mouse models,GPNMB expression was upregulated in the liver and secreted GPNMB levels were higher as well[148].Secreted GPNMB stimulates lipogenesisin vitroandin vivo[147].A neutralizing antibody targeting GPNMB reduces obesity and improves insulin sensitivity[147].In both mice and humans,GPNMB levels are positively correlated with obesity and insulin resistance[147].GPNMB is thus a promising therapeutic target for treatments of obesity and diabetes.
CONCLUSION
The liver has numerous endocrine functions such as direct hormone and hepatokine production,hormone metabolism,synthesis of binding proteins,and processing and redistribution of metabolic fuels.In the last 10 years,many new endocrine functions of the liver have been discovered(Figure 2).Several novel endocrine functions of the liver have been unraveled.The liver plays a key negative feedback regulatory role in the pancreatic α cell-liver axis which regulates pancreatic α cell mass,glucagon secretion,and circulating amino acid levels.Betatrophin and other hepatokines such as fetuin-A and FGF21 play important endocrine roles in modulating insulin sensitivity,lipid metabolism,and body fat weight.It is expected that more endocrine functions of the liver will be discovered in the near future.As endocrine function of the liver is a rapidly evolving field,controversial findings often exist;caution needs to be taken when interpreting novel findings to avoid over-simplification of complex metabolic processes and premature allocation of research resources.
杂志排行

World Journal of Hepatology的其它文章
- Incidence of umbilical vein catheter-associated thrombosis of the portal system:A systematic review and meta-analysis
- Role of endoscopic ultrasound in the field of hepatology:Recent advances and future trends
- Porta-caval fibrous connections—the lesser-known structure of intrahepatic connective-tissue framework:A unified view of liver extracellular matrix
- Promising diagnostic biomarkers of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis:From clinical proteomics to microbiome
- Fatty acid metabolism and acyl-CoA synthetases in the liver-gut axis
- Liver involvement in inflammatory bowel disease:What should the clinician know?