QuEChERS-HPLC法快速测定黄豆芽中6-苄基腺嘌呤含量的不确定度评定
2021-12-04刘飞波刘水平
刘飞波 - 王 希 刘水平 -
(株洲市食品药品检验所,湖南 株洲 412000)
6-苄基腺嘌呤是一种人工合成的生物生长调节剂,通过刺激细胞分裂引起植物生长和发育,抑制呼吸激酶而延长绿色蔬菜的保鲜,但长期过量食用会出现恶心、呕吐等现象[1-3]。2015年,国家食品药品监督管理总局、农业部、国家卫生和计划生育委员根据《中华人民共和国食品安全法》《中华人民共和国农产品质量安全法》等相关法律的规定,禁止豆芽生产过程中使用6-苄基腺嘌呤等物质[4]。
目前,GB/T 23381—2009规定的6-苄基腺嘌呤检测方法提取净化耗时较长,需要过SPE小柱处理,经济成本比较高。而QuEChERS法是一类快速、简便、经济、高效、耐用和安全的样品前处理方法,目前常用于植物源性产品中农药残留以及动物源性食品的检测。曾志杰等[5]公开发表了非标准方法液质方法测定豆芽中6-苄基腺嘌呤含量的合成不确定度,其前处理较为复杂,需要经过MCX固相小柱处理,成本较高。研究尝试结合QuEChERS前处理方法,采用高效液相色谱法检测果蔬类农产品中6-苄基腺嘌呤含量,根据CNAS-GL 06—2019《化学分析中不确定度的评估指南》和JJF 1059.1—2012《测量不确定度评定与表示》的要求,参考相关文献[5-11],通过分析各不确定度分量的来源,建立评定6-苄基腺嘌呤含量数学模型,计算分析过程,进行不确定度评定,利用不确定度评定对QuEChERS前处理果蔬类农产品的检测方法进行评价分析。
1 材料与方法
1.1 仪器与试剂
液相色谱仪:Thermo U3000型,美国赛默飞世尔科技公司;
电子天平:MS205DU型,瑞士梅特勒—托利多国际贸易有限公司。
6-苄基腺嘌呤:98.5%,德国Dr. Ehrenstorfer GmbH公司;
提取盐包、净化包:中国上海安谱实验科技股份有限公司;
甲醇:色谱纯,美国Honeywell公司;
甲酸:优级纯,上海国药集团化学试剂公司。
1.2 样品
果蔬类样品:黄豆芽4批,绿豆芽2批,草莓2批,黄瓜2批,市售。
1.3 试验方法
1.3.1 样品前处理 称取10.0 g均质试样于50 mL具塞离心管中,加入25.0 mL含体积分数1%甲酸的甲醇溶液,再加入提取盐包(内含4.0 g无水MgSO4、1.0 g无水醋酸钠),高速均质1 min,8 000 r/min离心5 min,取10 mL上清液于离心管中,加入净化包(内含有150 mg PSA及850 mg无水MgSO4,100 mg C18固相萃取吸附剂),涡旋1 min,8 000 r/min离心5 min,精确移取5.0 mL溶液至具塞离心管中,弱N2气流吹干,用甲醇定容至1.0 mL,甲醇定容后的溶液经0.45 μm微孔膜过滤,作为待测液供HPLC分析[7]。
1.3.2 色谱条件 色谱条件的选择参照GB/T 23381—2009以及相关文献资料[7],色谱柱:Welch Xtimate C18(4.6 mm×250 mm,5 μm);流动相:甲醇—0.02 mol/L乙酸铵混合液(V甲醇∶V乙酸铵=1∶1),等度洗脱;流速1.0 mL/min;柱温30 ℃;进样量20 μL;检测波长267 nm。
1.3.3 标准溶液的制备 标准溶液的配制参照GB/T 23381—2009 的要求,经高效液相色谱检测后,采用外标法定量。
(1) 标准储备液:精密称取6-苄基腺嘌呤对照品0.011 83 g置于10 mL容量瓶中,加入甲醇溶解并定容至刻度即得标准储备液1.165 mg/mL,于-18 ℃下保存。
(2) 标准工作液:用甲醇稀释上述标准储备液,配成5个不同质量浓度的系列标准工作液,分别为0.058 26,0.116 5,0.291 5,0.582 6,1.165 0 μg/mL,采用外标法进行定量。
1.3.4 6-苄基腺嘌呤含量计算
(1)
式中:
X——样品中6-苄基腺嘌呤含量,mg/kg;
C——测试溶液中的质量浓度,μg/mL;
C0——空白溶液中的质量浓度,μg/mL;
V——定容体积,mL;
m——样品取样量,g;
f——稀释倍数。
2 结果与分析
结合QuEChERS前处理方法,按“1.3.2项”色谱条件上机分析,6-苄基腺嘌呤与各杂质峰及溶剂峰能很好地分离,见图1。经过检测分析,1批黄豆芽有检出6-苄基腺嘌呤。以黄豆芽为主要分析对象,评价新的前处理方法对果蔬类农产品中6-苄基腺嘌呤含量测定的不确定度的影响。
2.1 不确定度来源分析

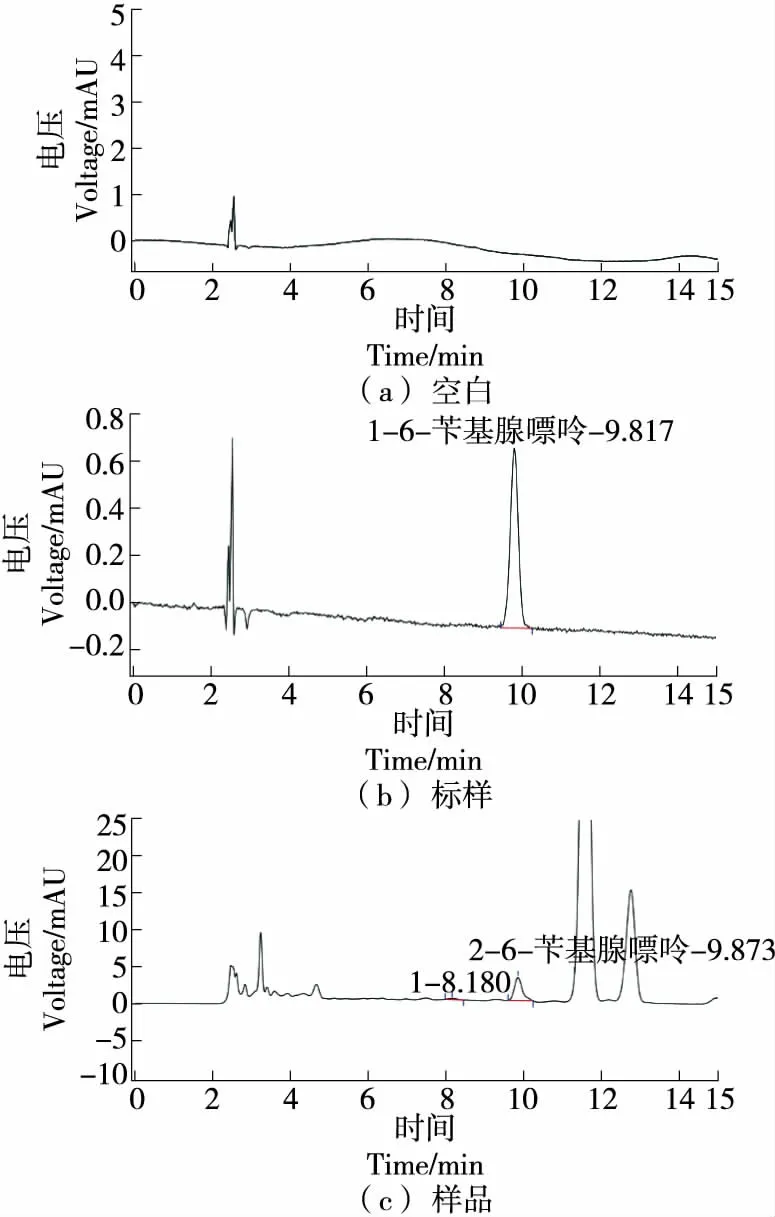
图1 6-苄基腺嘌呤样品和标样的HPLC分离效果图Figure 1 Separation of sample and 6-benzyladeninestandards by HPLC
2.2 不确定度量化分析
根据样品测定过程及数学模型,影响试验不确定度的主要分量见图2。

图2 不确定度影响因素构成图Figue 2 Constitution of uncertainty of influential factor
2.2.1 标准溶液配制过程中引入的相对标准不确定度
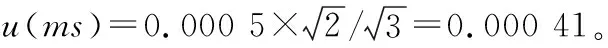
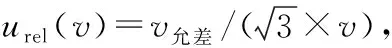
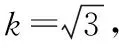
配制5个梯度的6-苄基腺嘌呤标准溶液(11.65 μg/mL→0.058 26 mg/mL)中使用1 mL的移液枪5次,10 mL A级容量瓶3次,100 mL A级容量瓶2次。引入的相对不确定度为:

综上,6-苄基腺嘌呤标准溶液配制过程中引入的相对标准不确定度合成为:
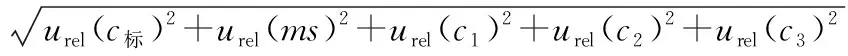

2.2.4 标准曲线引入的相对标准不确定度 6-苄基腺嘌呤和仪器响应峰面积见表2。试验将质量浓度为0.058 26,0.116 5,0.291 3,0.582 6,1.165 μg/mL的标准溶液分别进样3次,结果见表2。根据表2中的数据以浓度为横坐标,峰面积为纵坐标,拟合工作曲线方程为A=bC+d=0.792 7C-0.011 9(R2=0.998 2)。

表1 移液器引入的不确定度

表2 标准曲线拟合数据
称取10.24 g粉碎好的果蔬类(黄豆芽)样品,对同一样品进行5次测量,由校准曲线求得6-苄基腺嘌呤浓度为0.686 5 μg/mL,含量为0.335 mg/kg。校正曲线引入的相对标准不确定度urel(curve)的计算公式为:
urel(curve)=u(curve)/c,
(2)
(3)
(4)
式中:
b——斜率,0.797 2;
p——测试次数,5;
n——校准次数,15;
c——样品黄豆芽中的质量浓度,0.686 5 μg/mL;


Ai——第i个校准标准溶液的测定峰面积,mAu·min;
ci——第i个校准标准溶液的浓度,μg/mL;
d——标曲截距,-0.011 9。
SR——标准溶液峰面积残差的标准差,0.008 33。
将以上数据代入式(2)求得urel(curve)=0.028 2。
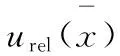
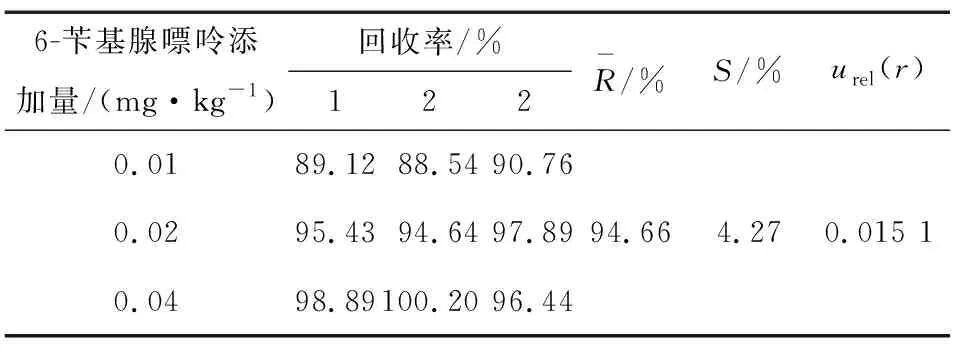
表3 回收率引入的不确定度
重复测定阳性样品(黄豆芽),平行测定6份,所得的峰面积按标曲换算成质量浓度,分别为0.335,0.331,0.339,0.336,0.333,0.326 mg/kg,平均质量浓度为0.334 mg/kg;测量重复性产生的不确定度为:
2.2.6 样品前处理定容引入的相对标准不确定度
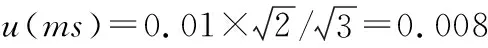

2.3 合成不确定度
综上分析,HPLC法测定果蔬类农产品中的6-苄基腺嘌呤的相对标准不确定度的分量见表4。
将合成相对标准不确定度为:
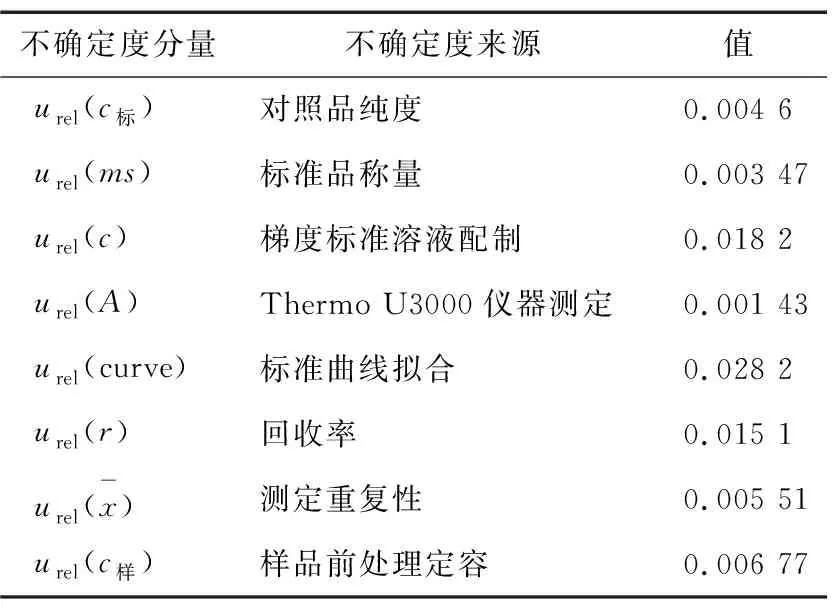
表4 相对标准不确定度分量表
取k=2,P=95%,则扩展相对标准不确定度U=k×uc=0.026 mg/kg,则果蔬类农产品(黄豆芽)中的6-苄基腺嘌呤含量可以表示为:(0.335±0.026) mg/kg,k=2(95%的置信区间)。即:6-苄基腺嘌呤的含量测定结果出现在(0.335±0.026) mg/kg区间的概率为95%。
由表4可见,文中不确定度的主要来源是标准曲线的拟合[urel(curve)=0.028 2]、梯度标准溶液配制[urel(c)=0.018 2]以及测定的回收率[urel(r)=0.015 1]。曾志杰等[5]有关液质方法测定豆芽中6-苄基腺嘌呤含量的合成相对标准不确定度uc=0.033 4,略低于文中的合成相对标准不确定度0.038 3,但其未考虑整个前处理定容所引入的不确定度,分析不够全面。唐韵熙[6]有关液质方法对豆芽中4-氯苯氧乙酸、6-苄基腺嘌呤残留量的不确定度评定,其合成相对标准不确定度urel=0.093 98,远高于文中的不确定度评定的数据,即使不考虑检测仪器引入的相对标准不确定度urel(fm)=0.04,其合成相对标准不确定度urel(x)=0.079 6,也高于文中的。唐韵熙[6]的研究中不确定度的主要来源分别是样品的前处理[urel(样品)=0.057 74]、拟合标准曲线[urel(curve)=0.040 113]、检测仪器[urel(A)=0.04]以及梯度标准溶液配制[urel(c)=0.030 16]。通过比较两者之间的不确定度的主要来源,作者发现复杂的前处理方式,引入了大量的不确定度。新的前处理方法简化了前处理步骤,减少了溶剂的消耗,有利于环境保护和检验人员的身体健康。
3 结论
对果蔬类农产品中6-苄基腺嘌呤进行检测时,采用QuEChERS法进行前处理,能够提升前处理速度,减少溶剂的消耗,减少污染,保护人员身体健康。样品的称取、提取、净化以及定容等过程都会引入不确定度,但其主要来源于拟合标准工作曲线求得样品浓度过程以及标准溶液的配制,而其他因素影响较小。因此,平时检验检测过程中要注意前处理的规范性,尽可能选用高精度的A级量具以及精确配制标准溶液等。同时应定期对高效液相色谱仪进行保养、期间核查以及检定校准,以保持仪器状态良好,降低仪器本身带来的不确定度的影响,保证测定结果的可靠性。
