高效液相色谱-示差折光检测器测定柴油中多环芳烃
2021-12-04周太云胡莉香中国石化油化工股份有限公司荆门分公司湖北荆门448002
周太云,胡莉香(中国石化油化工股份有限公司荆门分公司,湖北 荆门 448002)
0 引言
目前企业生产监控柴油中芳烃含量的主要方法有荧光指示剂法(FIA)、气质联用法(GC-MS)、高效液相色谱-示差折光检测器法(HPLC-RID)等。FIA仅适用于测定终馏点低于315 ℃的浅色石油馏分,生产企业柴油终馏点多数在360 ℃左右,一般呈黄绿色,所以该方法不适合测定柴油中的芳烃含量;GC-MS可以得到柴油详细的组成信息,但是需要将柴油预分离成饱和烃和芳烃后分别进行色谱分析,耗时耗力,不利用快速分析;HPLC-RID法选用单一模型化合物作为标准物进行定量,而柴油分子组成复杂性,不同结构的化合物的折射率存在一定的差异[1],从而对测定结果有一定的影响,但该法简便、快速,并且有良好的重复性和准确性[2],在车用柴油标准GB 19147中被列为仲裁检测方法[1]。文章对HPLC-RID法测定柴油单环芳烃、双环芳烃和三环+芳烃试验条件进行了优化,使用标准曲线法定量,结果达到预期的效果、符合标准要求。
1 实验部分
1.1 主要仪器和试剂
岛津 LC-20AT型高效液相色谱仪,配SPD-20AV检测器、SIL-20AC自动进样器、LC-20AD泵、CTO-20AC柱温箱及脱气单元、LabSolution 色谱工作站。正庚烷,HPLC级;环己烷:纯度大于99%;邻二甲苯、1-甲基萘:纯度大于98%;二苯并噻吩、9-甲基蒽:纯度大于95%。
1.2 仪器条件
色谱柱:ZORBAX NH2柱(4.6 mm×250 mm×5 μm);柱温:35 ℃;流动相:正庚烷,流量1.0 mL/min;检测器:35 ℃;进样量:10 μL。
1.3 标准溶液的制备
1.3.1 配制系统性能验证标准溶液(SPS)
称量1.0 g±0.1 g环己烷、0.5 g±0.05 g邻二甲苯、0.05 g± 0.005 g二苯并噻吩、0.05 g±0.005 g 9-甲基蒽,置于100 mL容量瓶中,用正庚烷稀释至刻度。二苯并噻吩、9-甲基蒽不易溶于环己烷和邻二甲苯中,可用超声波等方法使其充分溶解后再用正庚烷稀释。使用密封较好的瓶子,在-20℃的冰箱中保存备用。
1.3.2 标准溶液的制备
配制标准溶液A、B、C、D:按照如表1所示推荐的浓度称量标准物质(精确至0.1 mg),置于100 mL容量瓶中,用正庚烷稀释至刻度。
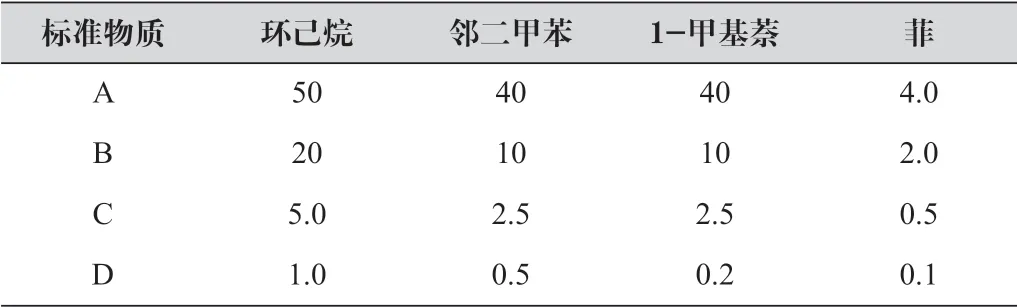
表1 标准溶液的浓度 单位:mg/mL
1.4 实验步骤
称取推荐量的柴油试样于10 mL容量瓶中,用正庚烷稀释至刻度,定量注入液相色谱系统中,色谱峰依次流出顺序为非芳烃、单环芳烃、双环芳烃和三环+芳烃。双环芳烃流出后,在预先测定的时间点使反冲洗阀反冲洗,以便把三环+芳烃洗脱成一尖锐的窄峰。使用示差折光检测器检测,外表标准曲线法定量。典型的样品色谱图如图1所示。
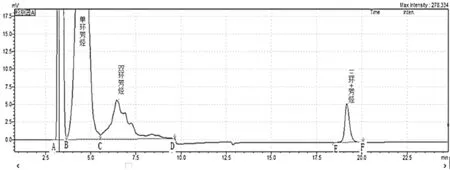
图1 柴油试样色谱图
2 结果与讨论
2.1 色谱柱的选择
柴油中烷烃是非极性物质,芳烃为极性物质,流动相使用的是有机溶剂正庚烷,与之配套色谱柱为极性氨基柱。对Waters Spherisorb® 5.0 μm NH2柱 和Agilent ZORBAX NH25.0 μm柱进行比较,发现Waters柱分离单环芳烃和双环芳烃时效果不好(如图2所示),导致多环芳烃分析结果误差大,而Agilent柱对单环芳烃和双环芳烃有良好的分离效果(如图1所示)。实验选择Agilent ZORBAX NH2柱作为分离柱。

图2 Waters柱分离情况色谱图
2.2 色谱柱流速的选择
柱流速影响柴油中各组分的分离度。SH/T 0806—2008建议最佳流速0.8~1.2 mL/min,试验考察了柱流速分别在1.0 mL/min、1.2 mL/min时各组分的分离情况。当流速为1.2 mL/min时双环芳烃以及三环+芳烃保留时间很近,不利于反吹时间的确定;当柱流速为1.0 mL/min时,单环芳烃、双环芳烃以及三环+芳烃可以得到有效的分离。故选择流动相的流速为1.0 mL/min。
2.3 柱温的选择
柱温对样品中各组分的分离效果有较大的影响,温度波动影响分析结果的重复性和准确性。在其他条件不变的情况下,考察不同柱温下样品的分离度,当柱温较低时,各组分间的分离度较好,但分析时间过长,半峰宽过大,峰形变差;当柱温较高时,分析时间较短,但各组分间分离度变差,经过实验,将柱温选择为35 ℃。
2.4 反冲洗时间的确定
Agilent柱对饱和烃、单环芳烃和双环芳烃有良好的分离效果,对三环及三环以上的芳烃分析时间长且不能得到很好的分离,方法中采取反冲洗出色谱柱得到三环+芳烃色谱峰方法来计算三环+芳烃含量。反冲洗时间对目标物的分析结果影响较大,当设定的反冲洗时间大于计算值时,会有部分的三环+芳烃切割到双环芳烃中;当设定的反冲洗时间小于计算值时,会有部分的双环芳烃切割到三环+芳烃中。曾有文献对反冲洗时间允差值进行了考察[7],结果表明,其允许变化范围为±0.2 min。所以必须定期用标准溶液检查仪器的稳定性,若计算出的反冲洗时间变化大于0.2 min,需重新用SPS溶液来确定反冲洗时间。试验采用由二苯并噻吩和9-甲基蒽的保留时间计算反冲 洗时间。在选定的色谱条件下分析SPS系统校准标准溶液。没有反冲洗的系统校正标准溶液SPS色谱图如图3所示,进行反冲洗后的SPS标准溶液色谱图如图4所示。

图3 系统校正标准溶液SPS色谱图

图4 反冲洗后的SPS标准溶液色谱图
2.5 标准曲线、检出限
2.5.1 标准曲线
将2.3.2中的标准溶液在选定的仪器条件下进行分析,测量各个标准物质的峰面积。用单环芳烃、双环芳烃和三环+芳烃标准物质的浓度对峰面积作图绘制工作曲线,如图5所示。由工作曲线可以看出,单环芳烃、双环芳烃和三环+芳烃的工作曲线的相关系数均为0.99999,单环芳烃、双环芳烃和三环+芳烃最小浓度分别为0.08 mg/mL、0.06 mg/mL、0.005 mg/mL,该方法具有良好的线性,且满足方法要求的相关系数和截距。

?
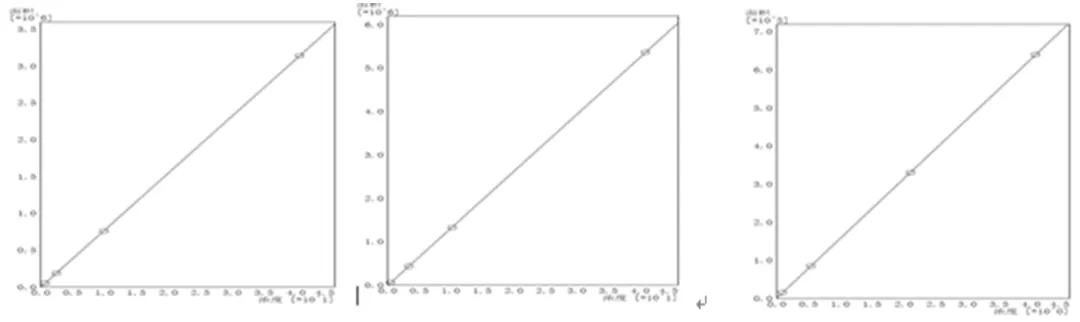
图5 标准物质工作曲线
2.5.2 检出限
色谱分析中一般选择3倍信噪比(S/N)所对应的浓度作为检出限,10倍信噪比所对应的浓度作为定量限,试验中将最低浓度的标准混合溶液逐级稀释,依次进样得到各组分的检出限和定量限,结果如表2所示。按取样量为1 g,稀释至10 mL时的样品各组分的检出限均为0.1%(质量分数),定量限均为0.2%(质量分数)。

表2 各组分的检出限测定
2.6 精密度和准确度
在样品中加入一定量的标准物质测定回收率,结果如表3所示,加标回收率在87.5%~108.7%之间。同时分析参考值为6.8% (质量分数)的柴油参考样,实际测定结果为6.4%(质量分数),满足参考样的重现性要求。由此可见,本方法的准确度较高。
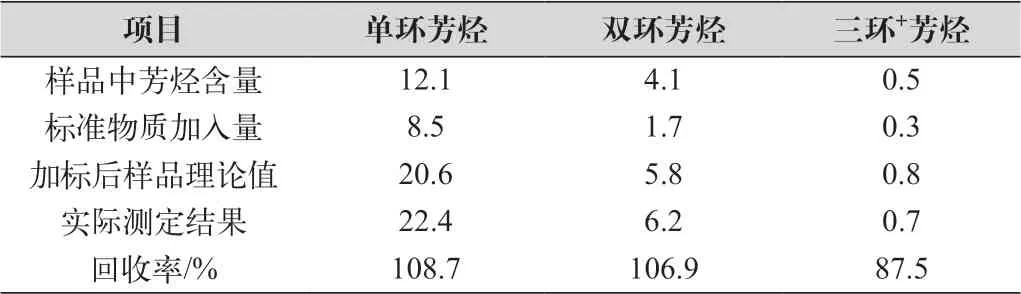
表3 加标回收结果 单位:%(质量分数)
本试验在选定的操作条件下,重复测定同一成品柴油和同一催化柴油,对同一样品测定10次的相对标准偏差在1.1%~8.5%,满足方法的精密度要求,同一样品多次测定的极差均小于SH/T 0806—2008的重复性要求,满足分析需要。
3 结语
(1)使用高效液相色谱-示差折光检测法测定柴油中芳烃及多环芳烃含量,该试验方法简便、快速,并且有良好的重复性和准确性,可在30 min之内完成一个样品的测试,应用范围广。
(2)对于不同芳烃含量的柴油馏分油可以通过增大或减小稀释倍数的方法进行检测,且液相色谱仪较气质联用仪便宜很多,在一定程度上节约了成本。
(3)反冲洗时间是该试验方法的一个重要参数,每分析5~10个样品或更换流动相后需要运行SPS标准样品来检查反冲洗时间是否合适,否则会引起测定误差。
