何首乌主要成分的肝毒性及其对药物代谢酶的影响
2021-11-24郑晓媛余志杰王璇周世文
郑晓媛 余志杰 王璇 周世文

中圖分类号 R285.5 文献标志码 A 文章编号 1001-0408(2021)21-2619-05
DOI 10.6039/j.issn.1001-0408.2021.21.10
摘 要 目的:研究何首乌主要成分的肝毒性,并基于代谢酶探讨其毒性机制。方法:采用ADMETlab 2.0平台预测何首乌的5种主要成分大黄素、大黄素甲醚、大黄酸、二苯乙烯苷、没食子酸对肝、皮肤、心脏等的毒性或致癌作用,并评估这些成分对细胞色素P450酶系(CYP1A2、CYP2C9、CYP2C19、CYP2D6、CYP3A4)的影响;检测不同浓度(10、20、40、80 μmol/L)的大黄素、大黄酸、二苯乙烯苷、没食子酸对人正常肝细胞L02存活率的影响;以胆红素为底物,采用体外反应体系考察何首乌主要成分对葡糖醛酸基转移酶1家族多肽A1(UGT1A1)活性的影响。结果:何首乌主要成分大黄素、大黄素甲醚、大黄酸、没食子酸对肝的毒性作用均较强。大黄素、大黄素甲醚对CYP1A2的抑制作用较强,对CYP2C9、CYP2D6、CYP3A4的抑制作用为中等;大黄酸对CYP1A2、CYP2C9的抑制作用为中等,二苯乙烯苷和没食子酸对上述各酶的抑制作用均较弱。大黄素(40、80 μmol/L)和没食子酸(40、80 μmol/L)均可显著降低L02细胞的存活率(P<0.01)。5、10、20、40、80 μmol/L的大黄素和没食子酸(5 μmol/L的大黄素除外)对UGT1A1酶的抑制率均显著升高(P<0.01),大黄素对UGT1A1酶的抑制作用为可逆的竞争性抑制。结论:何首乌主要成分大黄素、大黄酸、大黄素甲醚等具有肝毒性,其作用机制可能与抑制CYP1A2、CYP2C9活性以及竞争性抑制胆红素代谢限速酶UGT1A1的活性有关。
关键词 何首乌;大黄素;二苯乙烯苷;大黄酸;没食子酸;细胞色素P450酶;葡糖醛酸酶;肝毒性
ABSTRACT OBJECTIVE: To study the hepatotoxicity of main components of Polygonum multiflorum, and investigate its toxic mechanism based on metabolic enzymes. METHODS: ADMETlab 2.0 platform was used to forecast the toxic or carcinogenic effects of emodin, physcion, rhein, stilbene glycoside and gallic acid on liver, skin and heart. The effects of those components on cytochrome P450 enzyme system (CYP1A2, CYP2C9, CYP2C19, CYP2D6, CYP3A4) were evaluated. The effects of different concentrations of emodin, rhein, stilbene glycoside and gallic acid (10, 20, 40, 80 μmol/L) on the survival rate of normal hepatocyte L02 were detected. The effects of major components of P. multiflorum on the activity of UGT1A1 enzyme were studied by in vitro reaction system, using bilirubin as substrate. RESULTS: Main components of P. multiflorum, ie. emodin, physcion, rhein and gallic acid, showed strong toxic effects on the liver, while stilbene glycosides possessed weak toxic effects on the liver. Emodin and physcion had strong inhibitory effects on CYP1A2 and medium inhibitory effects on CYP2C9, CYP2D6 and CYP3A4; rhein showed medium inhibitory effects on CYP1A2 and CYP2C9, while stilbene glycoside and gallic acid possessed weak inhibitory effects on the above enzymes. Emodin (40, 80 μmol/L) and gallic acid (40, 80 μmol/L) could significantly reduce the survival rate of L02 cells (P<0.01). The inhibition rate of 5, 10, 20, 40, 80 μmol/L emodin and gallic acid (except for 5 μmol/L emodin) on UGT1A1 enzyme increased significantly (P<0.01), and the inhibition effect of emodin on UGT1A1 enzyme was reversible competitive inhibition. CONCLUSIONS: The main components of P. multiflorum, ie. emodin, rhein and physcion, are hepatotoxic; the mechanism of it may be associated with inhibiting the activity of CYP1A2 and CYP2C9 and competitively blocking rate-limiting enzyme UGT1A1 in the process of bilirubin metabolism.
KEYWORDS Polygonum multiflorum; Emodin; Stilbene glycosides; Rhein; Gallic acid; Cytochrome P450 enzyme; Glucuronidase; Hepatotoxicity
随着人类生活方式和使用中药的目的、方式发生改变,中药的安全性问题日益凸显,既往认为“补益养身”的何首乌及其制剂引起的肝损伤也屡见报道[1]。何首乌为蓼科植物何首乌Polygonum multliflorum Thunb.的干燥块根,具有抗衰老、降血脂、抗癌、抗炎、促进免疫调节、保护神经等作用[2];但也有相关研究发现,何首乌提取物可能导致肝毒性、肾毒性和胚胎毒性[3]。
研究发现,何首乌的主要成分有二苯乙烯苷、蒽醌类(包括大黄素、大黄酸、大黄素甲醚等)、鞣质(包括没食子酸等)和磷脂等,其中大黄素可通过刺激活性氧簇(ROS)的释放,启动外源性和内源性细胞凋亡途径来诱导HepaRG细胞凋亡[4-5];二苯乙烯苷、大黄素甲醚、大黄素-8-O-β-D-葡萄糖苷、顺式二苯乙烯苷、儿茶素等5种成分与何首乌的毒性具有较强的相关性[5],但具体作用机制尚不明确。
探讨药物对代谢酶的影响是药物肝毒性研究的重要组成部分。研究发现,药物在人体内的代谢途径可分为Ⅰ相代谢途径和Ⅱ相代谢途径,其中Ⅰ相代謝途径主要是氧化和羟基化,Ⅱ相代谢途径主要是硫酸酯化和葡萄糖醛酸化[6]。细胞色素 P450(CYP450)酶系是Ⅰ相代谢途径中的主要酶系,可影响多种药物代谢;葡萄糖醛酸转移酶(UGTs)是Ⅱ相代谢途径中的主要酶系,对胆红素和多种药物的代谢具有重要作用[6-7]。其中胆红素经血液循环至肝脏后,可通过UGTs形成胆红素葡萄糖醛酸化合物,进一步被排泄至胆囊及肠道进行清除;葡萄糖醛酸转移酶1家族多肽A1(UGT1A1)为该过程的限速酶,当其受到抑制时,可导致胆红素的清除减慢,从而表现出肝内胆汁淤积,进而损伤肝细胞[7]。
基于此,笔者首先采用ADMETlab 2.0平台预测何首乌主要成分(大黄素、大黄素甲醚、大黄酸、二苯乙烯苷、没食子酸)对肝、皮肤、心脏的毒性或致癌作用,并评估这些成分对细胞色素P450酶系的影响;采用人正常肝细胞L02验证何首乌主要成分的肝毒性,进而在体外反应体系层面研究何首乌主要成分对UGT1A1酶活性的影响,以期从代谢角度阐明何首乌肝毒性的作用机制。
1 材料
1.1 主要仪器
本研究所用主要仪器有Multiskan SkyHigh型全波长酶标仪、17R型高速低温离心机(美国Thermo Fisher Scientific公司),AE240 型电子天平(德国Sartorius公司),DK 600型电热恒温水槽 (上海精宏实验设备有限公司),F8型超细匀浆机(德国 Fluko公司) 。
1.2 主要药品与试剂
本研究所用大黄素(批号E7881)购自美国Sigma公司;大黄酸、二苯乙烯苷、没食子酸(批号分别为201812、201903、201909)均购自成都曼思特生物科技有限公司;RPMI1640培养基、胎牛血清(批号分别为31800022、10100147)均购自美国Gibco公司; CCK-8试剂盒(批号C0038)购自上海碧云天生物技术有限公司; HLA- UGT1A1酶(批号456411)购自美国Gentest公司;胆红素测定试剂盒(批号ab235627)购自英国Abcam公司;胆红素对照品(批号100077-201806,含量99.3%)购自中国食品药品检定研究院;其余试剂为实验室常用规格,水为纯化水。
1.3 细胞
人正常肝细胞L02购自中国科学院典型培养物保藏委员会细胞库。
2 方法
2.1 何首乌主要成分的毒性预测
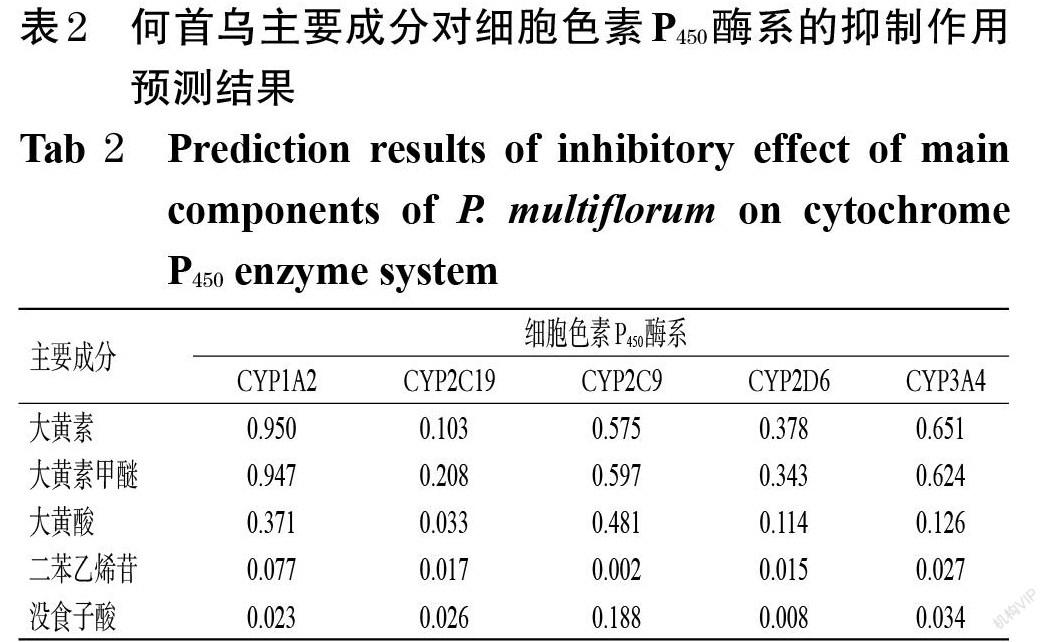
参考文献[8-9]的方法,采用ADMETlab 2.0平台(https://admet.scbdd.com/)进行预测。首先,将何首乌的5种主要成分大黄素、大黄素甲醚、大黄酸、二苯乙烯苷、没食子酸的化学结构式输入该平台,得出各成分的SMILE表达式;然后预测各成分对肝、皮肤、心脏、呼吸系统、芳烃受体的毒性作用及致癌作用,并评估这些成分对CYP450酶系(包括CYP1A2、CYP2C9、CYP2C19、CYP2D6、CYP3A4)的抑制作用。当评估预测值为0~<0.3时,提示成分的毒性/致癌/抑制作用弱;当该预测值为0.3~<0.7时,提示成分的毒性/致癌/抑制作用为中等;当该预测值为0.7~1.0 时,提示成分的毒性/致癌/抑制作用较强。
2.2 何首乌主要成分对L02细胞活性的影响考察
2.2.1 细胞培养 将L02细胞以含10%胎牛血清的DMEM培养基在37 ℃、5%CO2的恒温培养箱中培养。待细胞融合度达70%~80%时,以胰酶消化传代,进行后续实验。
2.2.2 细胞存活率的检测及形态学观察 取对数生长期的L02细胞,按5×103个/孔接种于96孔板中,然后分为大黄素不同浓度组(10、20、40、80 μmol/L,浓度参考文献[10]进行设置,下同)、大黄酸组不同浓度组(10、20、40、80 μmol/L)、二苯乙烯苷不同浓度组(10、20、40、80 μmol/L)、没食子酸不同浓度组(10、20、40、80 μmol/L),另设不加药物只加细胞的空白组,每组设5个复孔(大黄素甲醚与大黄素结构类似,故本实验不设置大黄素甲醚组)。待细胞贴壁后,各组细胞加入相应药物培养24 h,再加入CCK-8试剂10 μL培养1 h,然后采用酶标仪于450 nm波长下检测各孔的光密度值(OD),计算细胞存活率[细胞存活率=(OD空白组-OD实验组)/OD空白组×100%]。细胞存活率检测结束后,取各组细胞适量分别于显微镜下观察形态,并以大黄素不同浓度组细胞为代表拍照保存(其余组别的细胞显微图略)。
2.3 何首乌主要成分对UGT1A1酶活性的影响
2.3.1 UGT1A1酶抑制率的测定 参考文献[11-12]方法并进行改进,制备尿苷二磷酸葡萄糖醛酸(UDPGA)-缓冲系统,然后分为大黄素不同浓度组(5、10、20、40、80 μmol/L,浓度根据预实验结果设置,下同)、大黄酸不同浓度组(5、10、20、40、80 μmol/L)、没食子酸不同浓度组(5、10、20、40、80 μmol/L)以及空白组(加酶但不加药物),每组设5个复孔。各组均以胆红素(0.2 μg/μL)为反应底物,加入UGT1A1酶(0.5 U/L)1 μL以及相应药物,置于37 ℃恒温水浴条件下避光反应10 min后,加入冰乙腈-甲醇溶液600 μL终止反应;然后将反应液以15 000 r/min离心 25 min,取上清液175 μL加入96孔板中,并根据胆红素测定试剂盒说明书方法进行操作,采用酶标仪于550 nm波长处测定各孔OD值,并计算酶抑制率[酶抑制率=(OD空白组-OD实验组)/OD空白组×100%]。
2.3.2 大黄素对UGT1A1酶动力学的影响考察 以胆红素系列溶液 (0.125~2 mmol/L)为底物,参考“2.3.1”项下方法,测定不同浓度大黄素[0(作为空白对照)、20、40、80 μmol/L]对UGT1A1酶的抑制率,采用 Lineweave-Burk双倒数作图法确定大黄素对UGT1A1酶的反应类型[13]。
2.3.3 大黄素对UGT1A1酶抑制的可逆性考察 参考文献[14]方法,取UGT1A1酶溶液 (1 U/mL) 0.5 mL与 1 mg大黄素混合,于 37 ℃恒温水浴条件下预处理1 h,再置于4 ℃的 0. 1 mol /L 磷酸盐缓冲液(pH 6.8) 中透析24 h,透析期间每8 h更换1次磷酸盐缓冲液,作为大黄素透析组;同时,设置只加酶不加大黄素的空白透析组。另取UGT1A1酶溶液(1 U/mL) 0.5 mL与1 mg大黄素混合,不进行透析,直接置于4 ℃条件下保存 24 h 作为大黄素非透析组;同时,设定只加酶不加大黄素的空白非透析组。每组设5个平行。透析(或非透析放置)结束后,将混合溶液分别稀释10、50、100、500、1 000倍,測定OD值,并计算酶相对活性[酶相对活性=OD实验组/OD空白组×100%,式中透析或非透析实验组分别对应相应的透析或非透析空白组]。
2.4 统计学方法
采用SPSS 21.0统计软件进行分析。计量资料以x±s表示,多组间比较采用单因素方差分析。检验水准α=0.05。
3 结果
3.1 何首乌主要成分的毒性预测结果
3.1.1 毒性及致癌作用预测结果 何首乌中的大黄素、大黄素甲醚、大黄酸对肝、芳烃受体的毒性作用较强,二苯乙烯苷对皮肤、芳烃受体的毒性作用较强;没食子酸对肝、皮肤的毒性作用较强,详见表1。
3.1.2 酶抑制作用 大黄素、大黄素甲醚对CYP1A2的抑制作用较强,对CYP2C9、CYP2D6、CYP3A4的抑制作用中等;大黄酸对CYP1A2、CYP2C9的抑制作用中等,对CYP2D6、CYP3A4的抑制作用较弱;二苯乙烯苷和没食子酸对CYP1A2、CYP2C9、CYP2D6、CYP3A4的抑制作用均较弱;各成分对CYP2C19的抑制作用均较弱,详见表2。
3.2 何首乌主要成分对L02细胞存活率的影响
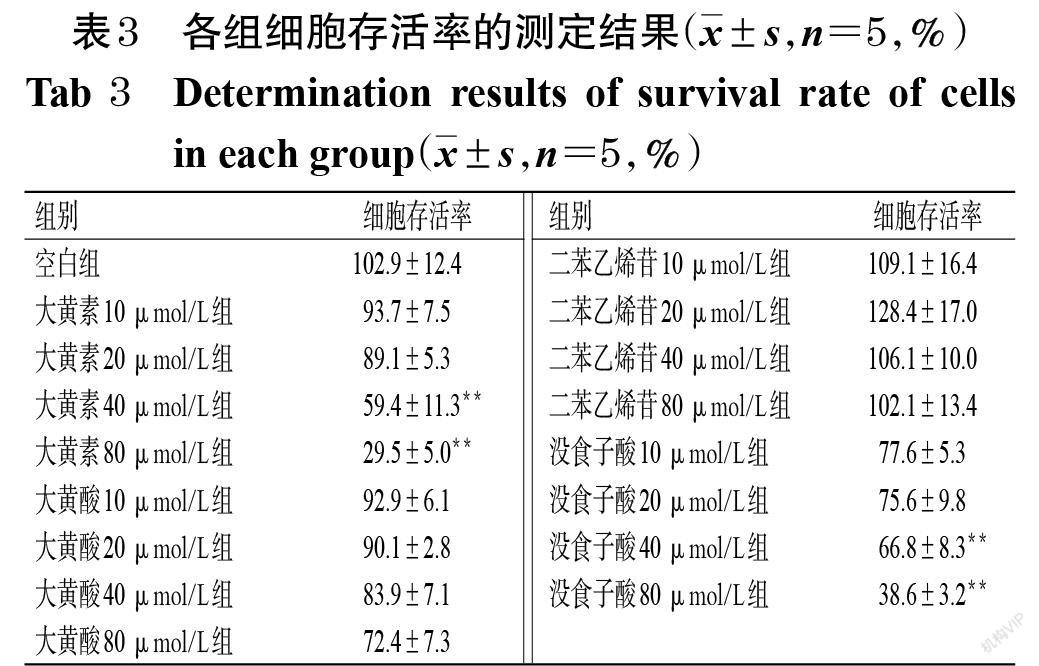
与空白组比较,大黄素40、80 μmol/L组和没食子酸40、80 μmol/L组细胞存活率均显著降低(P<0.01),且具有一定浓度依赖趋势;大黄酸不同浓度组和二苯乙烯苷不同浓度组细胞存活率差异无统计学意义,详见表3。细胞形态观察结果显示,空白组细胞形态正常;大黄素不同浓度组细胞随给药浓度的增加,表现出明显的色质凝集、核浓缩、核裂解等,且伴随发泡、凋亡小体生成,详见图1。
3.3 何首乌主要成分对UGT1A1酶活性的影响
3.3.1 UGT1A1酶抑制率 大黄素和没食子酸不同浓度组(大黄素5 μmol/L组除外)的UGT1A1酶抑制率较空白组均显著升高(P<0.05或P<0.01),大黄酸不同浓度组的UGT1A1酶抑制率较空白组无统计学意义,详见表4。
3.3.2 UGT1A1的酶动力学 大黄素对UGT1A1酶的抑制作用与给药浓度成正比,酶催化反应的最大反应速率(Vmax)以及米氏常数(Km)随给药浓度的升高而降低,表明大黄素对UGT1A1酶的抑制作用类型可能为竞争性抑制,详见图2。
3.3.3 UGT1A1酶抑制的可逆性 与空白透析组比较,空白非透析组的UGT1A1酶相对活性差异无统计学意义(P>0.05),表明透析对UGT1A1酶的清除无影响;与空白透析组比较,大黄素透析组稀释10、50倍后的UGT1A1酶相对活性均显著降低(P<0.01);与大黄素非透析组比较,大黄素透析组稀释10、50倍后的UGT1A1酶相对活性均显著降低(P<0.01),详见图3。
4 讨论
相关研究发现,患者短期或长期服用何首乌生品及其炮制品可发生肝毒性,严重者可发生肝衰竭[15]。英国、澳大利亚和加拿大等国的药品监督管理部门先后发布了何首乌致肝损伤的警告信息[16]。在我国,国家药物警戒平台收到的何首乌及其相关制剂的不良反应报告例次位居中药类前列,且多数以肝损伤为主;另外,国家药品监督管理局也屡次发布警示信息,提示何首乌及其相关制剂的肝损伤风险[17-19]。
计算机技术预测药物肝毒性是美国FDA推荐的常用方法[20],笔者在本文中采用计算机ADMETlab 2.0平台对何首乌的5种主要成分大黄素、大黄素甲醚、大黄酸、二苯乙烯苷、没食子酸的各器官毒性及致癌作用进行了模拟预测(由于药物致肝损伤可通过免疫介导途径损伤全身主要器官,故本文将皮肤、心脏、呼吸系统、芳烃受体均作预测分析[1])。结果显示,大黄素、大黄素甲醚、大黄酸、没食子酸对肝的毒性作用均较强。进一步通过细胞实验也发现,大黄素(40、80 μmol/L)和没食子酸(40、80 μmol/L)均可显著降低L02细胞的存活率。此外,大黄素、大黄素甲醚对CYP1A2具有较强的抑制作用,对CYP2C9、CYP2D6、CYP3A4具有中等抑制作用,这与文献研究结果基本一致[21]。由此推测,大黄素、大黄酸、大黄素甲醚等抑制细胞色素P450酶系的作用可能是何首乌主要成分导致药物性肝损伤的机制之一。
何首乌引发的肝毒性多伴有胆红素异常升高的表现,胆红素的代谢与Ⅱ相代谢途径中的葡萄糖醛酸结合环节有关,而在葡萄糖醛酸酶的1A1、1A3、1A4、1A5、1A6、1A7、1A8、1A9、1A10等多个异构体中,代谢胆红素的酶主要是UGT1A1酶[22]。基于此,本研究以胆红素为底物,从体外反应体系层面研究何首乌主要成分对UGT1A1酶活性的影响。结果显示,何首乌主要成分大黄素和没食子酸(5 μmol/L的大黄素除外)对UGT1A1酶的抑制率均显著升高;进一步通过酶动力学实验和透析实验发现,大黄素对UGT1A1酶的抑制作用为可逆的竞争性抑制。由此笔者推测,何首乌主要成分大黄素可通过抑制Ⅱ相代谢途径中的UGT1A1酶活性,从而增加胆红素代谢时间;另外,由于该抑制作用是可逆的竞争性抑制,故笔者推测在停止给药后,UGT1A1酶活性可逐渐恢复。
综上所述,何首乌主要成分大黄素、大黄酸、大黄素甲醚、没食子酸等具有肝毒性,其作用机制可能与抑制CYP1A2、CYP2C9活性以及竞争性抑制胆红素代谢限速酶UGT1A1的活性有關。
参考文献
[ 1 ] LIU Y,WANG W P,SUN M Y,et al. Polygonum multiflorum-induced liver injury:clinical characteristics,risk factors,material basis,action mechanism and current challenges[J]. Front Pharmacol,2019,10 (13):1467.
[ 2 ] 刘芳,李波.药物引起肝脏损伤的评价与检测[J].药物分析杂志,2010,30(10):1981-1984.
[ 3 ] 任红微,魏静,高秀梅,等.何首乌及其主要化学成分药理作用及机制研究进展[J].药物评价研究,2018,41(7):1357-1362.
[ 4 ] ZHENG X Y,YANG S M,ZHANG R,et al. Emodin-induced autophagy against cell apoptosis through the PI3K/AKT/mTOR pathway in human hepatocytes[J]. Drug Des Devel Ther,2019,13(9):3171-3180.
[ 5 ] DONG X X,FU J,YIN X B,et al. Induction of apoptosis in HepaRG cell line by aloe-emodin through generation of reactive oxygen species and the mitochondrial pathway[J].Cell Physiol Biochem,2017,42(2):685-696.
[ 6 ] K?HLE C,BOCK K W. Coordinate regulation of phaseⅠandⅡxenobiotic metabolisms by the Ah receptor and Nrf2[J]. Biochem Pharmacol,2007,73(12):1853-1862.
[ 7 ] VITEK L,OSTROW J. Bilirubin chemistry and metabolism;harmful and protective aspects[J]. Curr Pharm Des,2009,15(25):2869-2883.
[ 8 ] DONG J,WANG N N,YAO Z J,et al. ADMETlab:a platform for systematic ADMET evaluation based on a comprehensively collected ADMET database[J]. J Cheminform,2018,10(1):29.
[ 9 ] SHOOMBUATONG W,PRATHIPATI P,PRACHAYASITTIKUL V,et al. Towards predicting the cytochrome P450 modulation:from QSAR to proteochemometric modeling current drug metabolism[J]. Curr Drug Amtab,2017,18(6):540- 555.
[10] ZHANG H M,WANG F,QIU Y ,et al. Emodin inhibits coxsackievirus B3 replication via multiple signalling cascades leading to suppression of translation[J]. Biochem J,2016,473(4):473-485.
[11] 汪祺,张玉杰,戴忠,等.微粒体体系中胆红素及其代谢产物测定方法的建立[J].药物分析杂志,2015,35(9):1544-1550.
[12] 汪祺,戴忠,张玉杰,等.基于二相代谢酶介导胆红素代谢考察不同体系UGT1A1酶动力学参数[J].中国药学杂志,2015,50(19):1709-1714.
[13] FUJIMORI Y,WAKUI M,KATAGIRI H,et al. Evaluation of anticoagulant effects of direct thrombin inhibitors,dabigatran and argatroban,based on the lineweaver-burk plot applied to the clauss assay[J]. J Clin Pathol,2016,69(4):370-372.
[14] CHOWDHURY N R,ARIAS I M,LEDERSTEIN M,et al. Substrates and products of purified rat liver bilirubin UDP glucuronosyltransferase[J]. Hepatology,1986,10(6):123- 128.
[15] National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK). Shou wu pian (polygonum multiflorum) in the livertox[EB/OL]. (2019-09-25)[2021-07-12]. https://www.livertox.nih.gov/ShouWuPian.htm.
[16] 英国药品与保健品管理局.英国MHRA 警告何首乌的肝损害不良反应[J].中国药物警戒,2006,3(5):313.
[17] 国家食品药品监督管理总局.食品药品监管总局办公厅关于修订养血生发胶囊等6个含何首乌中药口服制剂说明书的通知[EB/OL].(2013-10-23) [2021-07-12]. http://samr.cfda.gov.cn/WS01/CL1706/93676.html.
[18] 国家食品药品监督管理总局.食品药品监管总局办公厅关于加强含何首乌保健食品监管有关规定的通知[EB/OL].(2014-07-09)[2021-07-12]. http://www.sda.gov.cn/WS01/ CL0847/102806.html.
[19] 國家食品药品监督管理总局. 国家食品药品监管总局提示关注口服何首乌肝损伤风险[EB/OL]. (2014-07-16) [2021-07-12]. http://www.sda.gov.cn/WS01/CL0051/102902.html.
[20] LIU Y,MAPA M S T,SPRANDO R L. Anthraquinones inhibit cytochromes P450 enzyme activity in silico and in vitro[J]. J Appl Toxicol,2021,41(9):1438-1445.
[21] XIONG G,WU Z,YI J,et al. ADMETlab 2.0:an integra- ted online platform for accurate and comprehensive predictions of ADMET properties[J]. Nucleic Acids Res,2021,49(W1):W5-W14.
[22] 常乙玲,李娜.何首乌及其制剂致药物性肝损伤系统性回顾研究[J].中国药业,2020,29(19):81-86.
(收稿日期:2021-09-10 修回日期:2021-10-14)
(编辑:唐晓莲)
