ELOVL1基因变异致IKSHD1例临床及基因分析
2021-11-18张广宇陈功勋李三松王明梅朱登纳
杨 磊 张广宇 陈功勋 李三松 王明梅 朱登纳
郑州大学第三附属医院儿童康复科(河南郑州 450052)
鱼鳞病样角化症、痉挛、髓鞘形成障碍和面部畸形(ichthyotic keratoderma、spasticity、hypomyelination and dysmorphic facies,IKSHD,OMIM:618527)是一种罕见的常染色体显性遗传病,由超长链脂肪酸延伸酶1(elongase of very long chain fatty acids 1,ELOVL 1)基因变异引起,于2018 年首次报道[1]。IKSHD 临床表现包括鱼鳞病样表皮增生和角质化增加、痉挛性截瘫、眼球震颤、构音障碍和语速减慢,面部畸形,伴有视神经萎缩可有视野和视力下降,头颅磁共振成像(MRI)表现为髓鞘化障碍[1-2]。搜索中国知网、万方、维普等数据库,国内尚无此病报道。现报告1例ELOVL1基因变异所致IKSHD病例并结合文献分析其基因致病性。
1 临床资料
患儿,男,1岁7个月,因不能独站、双下肢肌张力高伴皮肤脱屑,于2019年9月至郑州大学第三附属医院儿童康复科就诊。患儿9 月龄不能独坐、下肢支撑差伴肌张力增高,在当地医院诊断为“脑性瘫痪(痉挛型双瘫)”,接受康复治疗疗效差。患儿系G2P2,足月,因“瘢痕子宫”剖宫产娩出,出生体质量5.3 kg,无宫内窘迫及生后窒息史,Apgar 评分10 分。母孕期合并妊娠期糖尿病,皮下注射胰岛素治疗后血糖控制在正常范围,否认毒物及射线接触史。父亲体健,否认近亲结婚;姐姐5岁,体健。体格检查:身长84 cm(P50~85),体质量14 kg(>P97),头围48 cm(P50~85);神志清,精神可;肘部、膝部、小腿背侧及踝部皮肤干燥,上覆糠秕样鳞屑,局部皲裂、色素沉着;面容异常,眼距宽、大耳、长人中;双眼球向右斜视、水平震颤,双侧瞳孔等大等圆,对光反射灵敏;心、肺、腹未见明显异常;能完成简单指令,认识五官,会简单模仿;语言表达能力落后,发音不清;双手能主动抓物,精细动作欠佳;短暂拱背坐、稳定性差,不能独站,扶立位双下肢能部分支撑体重、双下肢交叉、尖足;双上肢肌张力正常,双下肢肌张力高,改良Ashworth量表(Modified Ashworth Scale,MAS)分级3级;双侧膝腱反射亢进,踝阵挛阳性。实验室检查:血常规、肝功能、肾功能、心肌酶、甲状腺功能、血氨基酸及尿有机酸检测均未见异常。脑电图未见异常。听觉诱发电位示双耳高频听阈60 dB。头颅MRI 示脑白质髓鞘化障碍(图1)。脊髓MRI 未见异常。肝、胆、胰、脾、双肾、膀胱、输尿管彩超未见异常。言语评估示语言发育迟缓、构音障碍。0~6岁儿童神经心理发育评定示发育商大运动30.3,精细动作75.8,适应能力83.3,语言60.6,社交 行为58.1。
为进一步明确诊断,经医院医学伦理委员会审查通过及家长知情同意,分别取患儿及父母外周血3 mL行全外显子检测和Sanger测序验证。结果患儿1号染色体上的ELOVL 1基因存在c.464 G>C 错义变异,此序列变化为ELOVL1基因第464位核苷酸鸟嘌呤突变为胞嘧啶,导致其蛋白质翻译在第155 位甘氨酸变成丙氨酸(p.Gly 155 Ala),家系验证结果示父母均未见此变异(图2)。该变异为新发变异,且无家族史(PS2);ESP数据库、千人数据库、EXAC数据库中正常对照人群中未发现的变异(PM2);生物信息学软件Clinpred、LRT、Mutationtaster、Provean、PolyPhen2等对蛋白功能预测为有害变异。根据ACMG指南,c.464G>C位点致病性等级为可能致病变异(PS2+PM2)。患儿临床表型及头颅MRI 表现高度符合IKSHD,综合判定ELOVL 1基因的c.464 G>C 变异为患儿的致病原因,诊断为IKSHD。
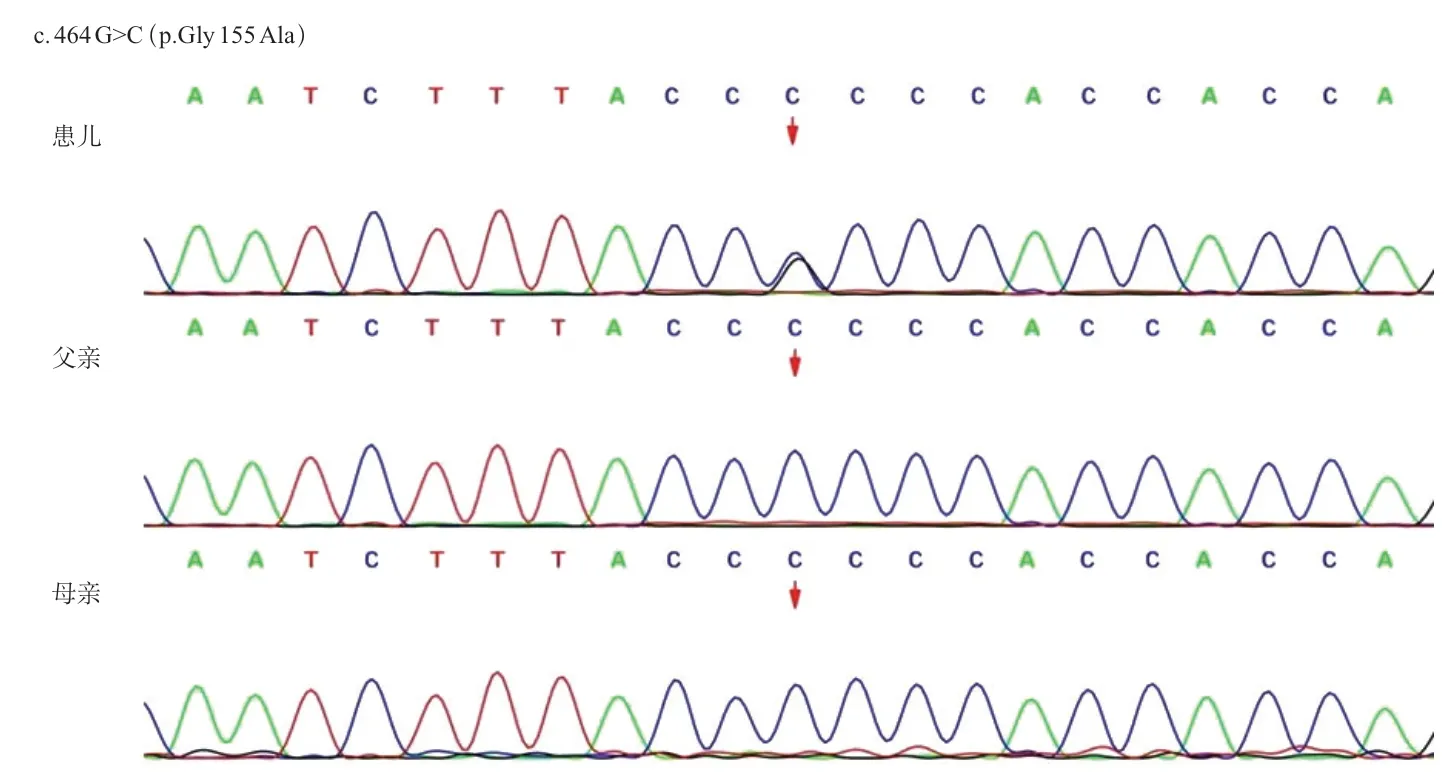
图2 患儿及其父母ELOVL1基因测序图
入院后给予患儿运动、手作业治疗及听觉语言训练等综合康复治疗,皮损处保持清洁、维生素E 乳膏外用,治疗1个月后返回当地医院继续康复治疗。2岁4月龄复查患儿头颅MRI较前无明显变化,仍为白质髓鞘化障碍(图1)。2 岁11 月龄随访,患儿皮损处皮肤无明显变化,智力正常,双手精细动作欠佳,独坐稳健,能抓站,不能扶物走,双下肢肌张力仍明显增高。MAS分级3级,复查0~6岁儿童神经心理发育评定示发育商大运动35.1,精细动作82.6,适应能力90.1,语言78.4,社交行为75.9。
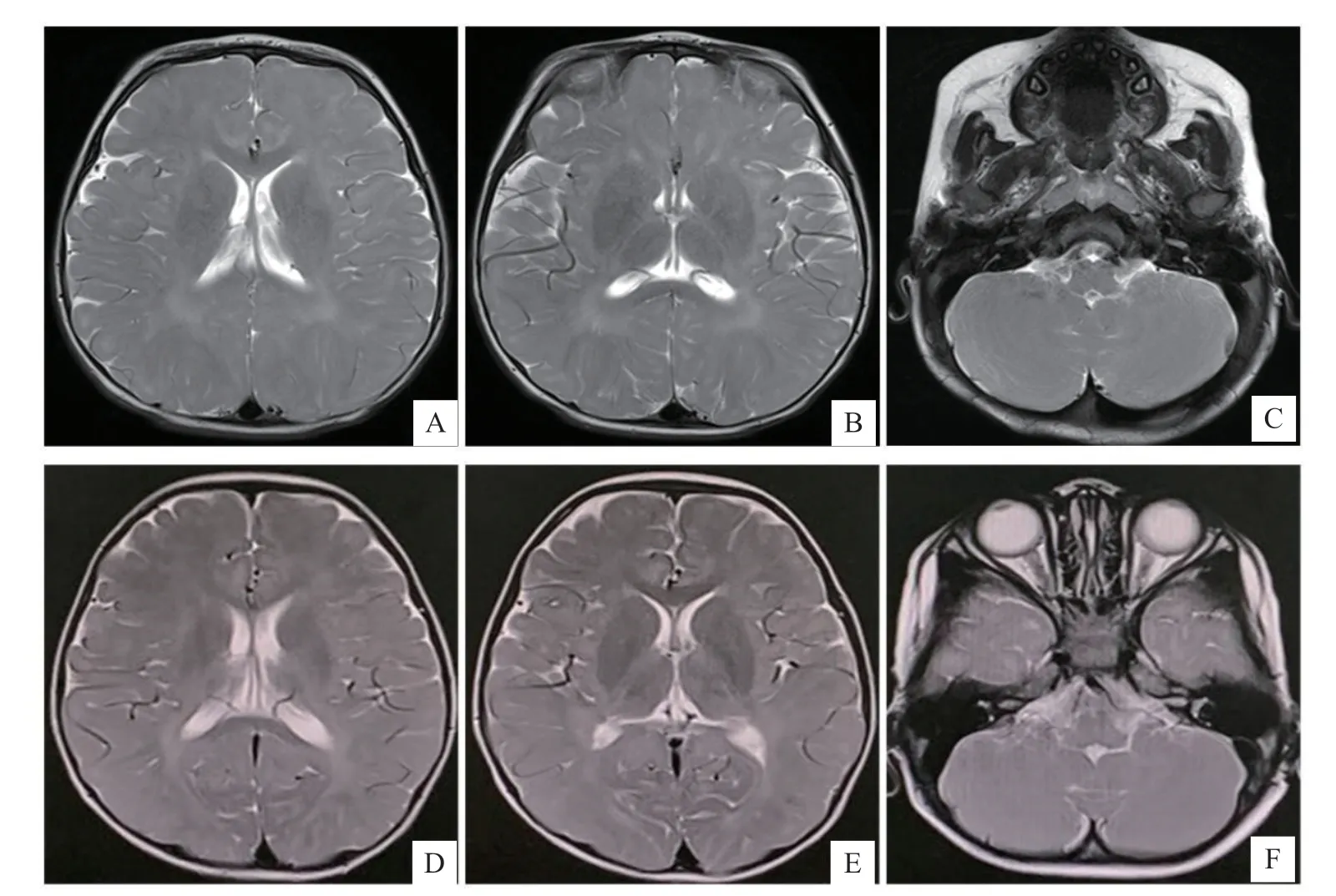
图1 患儿头颅MRI(平扫T2 加权像)
2 讨论
IKSHD 是一种以鱼鳞病样角化症、痉挛性截瘫、脑白质髓鞘化障碍、面部畸形等为主要表现的多器官受累的疾病。2018 年首次报道2例无任何血缘关系的波兰患儿同时出现鱼鳞病样角化症、痉挛性截瘫、脑白质髓鞘化障碍、婴儿期眼球震颤、面部畸形特征等显著体征,通过进一步遗传和功能研究证实为ELOVL1基因变异(c.494C>T,p.Ser165Phe)引起的新的遗传病[1]。IKSHD 的临床表现与ELOVL 1功能改变所受累器官密切相关,国外报道的2例患儿临床表现包括鱼鳞病样表皮增生和角质化增加、痉挛性截瘫、眼球震颤、构音障碍和语速减慢,视野缺损和视力下降,面部轻度畸形(长鼻子、球茎顶、嘴唇丰满、长人中,眉毛宽,大耳),头颅MRI 表现为髓鞘化障碍,听力检查为双耳高频感音神经性听力损失,眼底检查可有视神经萎缩表现[1-2]。
本例患儿存在IKSHD 的典型临床表现,包括肘膝踝关节及小腿背侧皮肤鱼鳞病样改变,下肢痉挛性截瘫,眼球震颤伴斜视,构音障碍,面部轻度畸形特征等;头颅MRI示脑白质髓鞘化障碍;听力检查示感音神经性听力损伤,高频听阈60 dB。全外显子检测发现患儿ELOVL1基因杂合变异c.464G>C(p.Gly155Ala),为新发变异,家系验证患儿父母均为野生型,符合常染色体显性遗传规律,且ESP 数据库、千人数据库、EXAC数据库中正常对照人群中未发现该变异,根据ACMG 指南评级为可能致病变异。依据患儿临床表现、辅助检查及基因检测结果,诊断为IKSHD,且为不同于国外报道的新的变异位点。
ELOVL1基因位于染色体1p34.2,由8个外显子组成,编码蛋白为ELOVL1,该蛋白含有279个氨基酸。ELOVL 1 是一种催化超长链脂肪酸(very long-chain fatty acid,VLCFA)合成中限速步骤的延伸酶,在中枢神经系统的髓鞘部分和皮肤、肾脏、胃、肺等组成性表达,对神经和表皮屏障功能非常重要[3-4]。ELOVL1在合成含有20 个或更多碳(C 20:0、C 22:0 等)的饱和及不饱和脂肪酸中起作用,在合成C 24:0-C 28:0和C 26:1 中具有特别强的活性[5]。国外多篇研究报道了ELVOL1的作用,如在ELOVL1基因缺陷的小鼠中发现该基因编码负责产生C 20-C 28 脂肪酸的延伸 酶[6];在X 连锁肾上腺脑白质营养不良(X-ALD)患者的成纤维细胞中,ELOVL1基因敲除导致C26:0水平显著降低,ELOVL1在C24:0和C26:0合成中的作用也从其药理抑制的结果中得到证实,苯扎贝特可通过抑制ELOVL1减少X-ALD成纤维细胞中VLCFA的 积累[7]。
ELOVL1基因在大脑和皮肤中高表达,基因变异致ELOVL 1 酶活性降低,导致其合成产物VLCFA 的缺乏。VLCFA是表皮脂质层的基本组成部分,是表皮角质层角质细胞之间的疏水结构,构成皮肤通透性屏障,有报道ELOVL1基因敲除小鼠出生后不久即因鱼鳞病样症状表皮屏障缺损而死亡[6],患儿皮肤样本脂质分析发现C 26-神经酰胺和鞘磷脂减少,从而出现鱼鳞病样皮肤改变。VLCFA 亦是诸如鞘脂和磷脂等复杂脂质的重要组成部分,神经元的发育和髓鞘的形成主要依赖于VLCFA的有效性和对其合成及降解的严格控制[7],白质髓鞘化障碍可导致痉挛性截瘫及眼球震颤,视神经萎缩可导致视野缺损及视力下降。生化分析结果亦发现p.Ser165Phe变异会导致C20:0和C 22:0 的积聚,尽管其不具毒性,但不能排除变异的ELOVL1引起某些其他脂质化合物蓄积的情况,这些化合物可能对神经元和表皮细胞有害[1]。由于报道较少,蛋白分子功能改变机制方面尚不清楚。ELOVL 1变异体Ser 165 及其侧翼氨基酸在整个脊椎动物中高度保守,并被预测位于内质网腔和跨膜螺旋5 之间的边界,这与导致脊髓小脑共济失调的位于内质网腔和螺旋7 交界处的ELOVL 4(p.W 246 G)和ELOVL 5(p.G 230 V)已知显性错义变异相符[2,8]。显性遗传模式表明单倍体不足或获得病理功能是潜在的病理机制,但此基因错义变异无法用单倍体不足解释,获得病理功能的可能性更大,需要进一步研究证实。国外报道2例患儿变异位点为c.494 C>T,本例患儿ELOVL1基因变异位点与上述位点不同,临床表现方面,本例患儿鱼鳞病样皮肤范围主要位于四肢,胸腹部皮损不明显,较国外2例患儿皮损程度略轻,可能与不同位点变异导致的酶功能降低程度相关,可通过进一步分子学及酶学研究其致病机制。
IKSHD 的提出丰富了遗传性鱼鳞病及髓鞘形成障碍性疾病的疾病谱。遗传性鱼鳞病是一组由影响表皮功能的基因变异导致的单基因遗传病,同时也可能是伴随皮肤外症状的综合征,例如伴发神经系统症状的Sjogren-Larsson综合征,伴发肌病的钱林-多尔夫曼综合征,由GJB2基因变异引起的角膜炎、鱼鳞病、耳聋综合征等[9-10]。而中枢神经系统髓鞘形成障碍性疾病是一种具有不同神经系统表现的疾病,在头颅MRI上显示髓鞘形成障碍,与多种具有不同功能的基因变异以及染色体畸变有关,如佩梅病(PLP1基因)、佩梅样病(GJA12/GJC2基因)、18q综合征等[11]。曾报道一种伴有鱼鳞病的髓鞘形成障碍性神经系统疾病,鱼鳞病、痉挛型四肢瘫和精神发育迟滞,是由延长酶ELOVL4的隐性变异引起[12],IKSHD与其区别主要在于患儿智力发育基本正常。
目前IKSHD尚无特效治疗方法,以对症治疗和康复训练为主,其预后需长期随访观察。本例患儿存在运动障碍、姿势反射发育异常、下肢肌张力增高等症状,初期诊断为脑性瘫痪,6 月龄时MRI 报告未见明显异常,围生期无明显高危因素,查体发现局部皮肤鱼鳞病样改变,复查头颅MRI 发现髓鞘化障碍,血氨基酸及尿有机酸检测均未见异常,经全外显子检测明确患儿诊断。
综上,IKSHD 临床表现为鱼鳞病样角化症、痉挛性截瘫及脑白质髓鞘形成障碍等,其致病原因为ELVOL 1基因变异。本例IKSHD 患儿通过全外显子测序及Sanger 测序验证,检测到ELVOL 1基因变异c.464 G>C,为新发现变异,拓宽了IKSHD 致病基因的变异谱。对临床中无明显高危因素或伴发其他症状的疑似脑性瘫痪患儿应仔细查体,完善头颅MRI等辅助检查,必要时行基因检测明确病因,对于遗传咨询及优生优育具有重要意义。
