HPLC 法测定依帕司他片清洁验证中残留物
2021-11-17尤锐薛雯蔚冯小雪扬子江药业集团南京海陵药业有限公司江苏南京210049
尤锐,薛雯蔚,冯小雪(扬子江药业集团南京海陵药业有限公司,江苏 南京 210049)
0 引言
糖尿病是一种常见的慢性非传染性疾病,已成为严重的世界性公共卫生问题。《国务院关于实施健康中国行动的意见》[1]指出,我国是糖尿病患病率增长最快的国家之一。据国际糖尿病联盟(IDF) 数据显示,截至2019 年,在20 岁到79 岁的人群中,中国糖尿病患者数总人数约为1.164亿[2]。然而,糖尿病致死原因主要归于其并发症。据世界卫生组织统计,糖尿病并发症高达100 多种,是目前已知并发症最多的一种疾病。糖尿病死亡者有一半以上是心脑血管所致,10% 是肾病变所致。因糖尿病截肢的患者是非糖尿病的10~20 倍。临床数据显示,糖尿病发病后10 年左右,将有30%~40% 的患者至少会发生一种并发症,且并发症一旦产生,药物治疗很难逆转,因此强调尽早预防糖尿病并发症。糖尿病周围神经病变(diabetic peripheral neuropathy, DPN) 属于糖尿病微血管并发症,发病机制较为复杂,是老年2 型糖尿病患者反复住院的重要危险因素[3]。依帕司他片主要用于治疗糖尿病周围神经病变,是预防糖尿病并发症的常用药物。临床研究证明[4],依帕司他联合甲钴胺治疗糖尿病周围神经病变效果显著,可改善神经传导速度与临床表现,且患者无明显不良反应,用药安全性较高。
最大限度地降低药品生产过程中污染、交叉污染是药品生产质量管理的核心内容之一[5]。出于生产成本、生产空间的局限性,做到降本增效,在药品生产过程中,不同药品共用生产线和生产设备的情况不可避免。制定有效、便捷的清洁程序是防止污染、交叉污染的重要举措之一。清洁验证[6]是证明清洁程序能有效清洁设备并满足其预定用途的直接证据。基于此,本文参照《日本药典》中依帕司他含量测定的检测方法,优化实验条件,建立高效液相色谱法测定依帕司他片清洁验证中的化学残留。
1 实验部分
1.1 主要仪器与试剂
Agilent 1260 高效液相色谱仪(安捷伦科技有限公司);XPE205DR 分析天平(梅特勒-托利多)。
磷酸二氢钾(分析纯,国药集团化学试剂有限公司);磷酸氢二钠(分析纯,国药集团化学试剂有限公司);乙腈(色谱纯,TEDIA);乙醇(色谱纯,TEDIA);N, N-二甲基甲酰胺(色谱纯,TEDIA);布签(TEXWIPE)。
1.2 实验方法
1.2.1 色谱条件色谱柱:十八烷基硅烷键合硅胶为填充剂(Waters Xbrige C18 150mm×4.6 mm,5 μm);流动相:磷酸盐缓冲液[0.05 mol/L磷酸二氢钾溶液,用0.05 mol/L 磷酸氢二钠溶液调节pH 值至6.5(0.05 mol/L 磷酸二氢钾溶液与0.05 mol/L 磷酸氢二钠溶液的体积比约为200∶85)]-乙腈(65∶35);检测波长:294 nm;柱温:25 ℃;进样盘温度:4 ℃;进样量:50 μL;流速:1.0 mL/min;运行时间:10 min;洗针方式:冲洗端口,15 s;洗针溶液:乙腈。
1.2.2 标准溶液的配制和标准曲线的绘制
精密称取依帕司他对照品约30.5 mg,置50 mL 容量瓶中,加N, N-二甲基甲酰胺适量,充分振摇使溶解,并稀释至刻度,摇匀;精密量取上述溶液2 mL,置100 mL 容量瓶中,加流动相稀释至刻度,摇匀,即得对照品贮备液。精密量取对照品贮备液5 mL 置50 mL 容量瓶中,加流动相稀释至刻度,摇匀,即得对照品溶液。
精密量取依帕司他对照品贮备液适量,使制成浓度范围在定量限至300% 浓度水平(约为3.66 μg/mL) 之间的6 个浓度水平的溶液。按1.2.1 项下色谱条件记录色谱图,以浓度(c) 为横坐标,峰面积(A) 为纵坐标,计算回归方程及相关系数。浓度级别及试验结果如表1 所示。
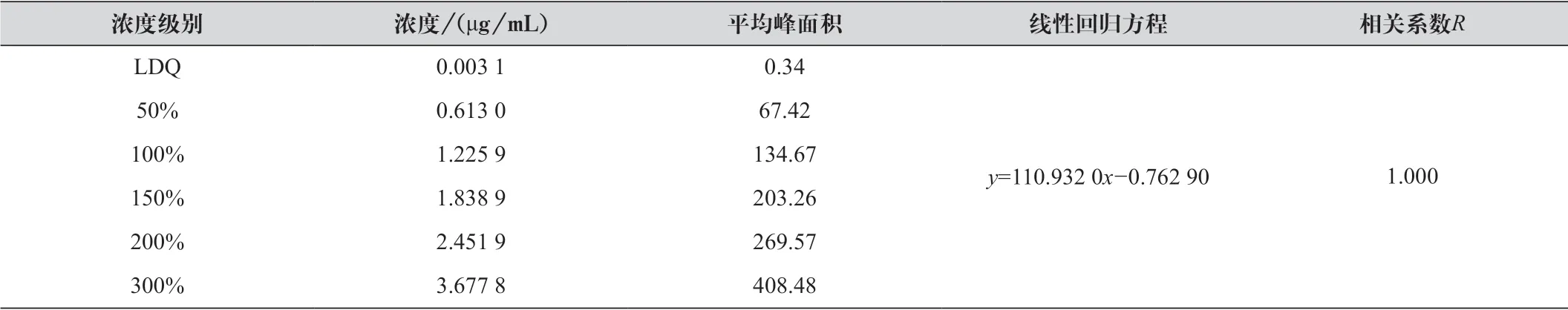
表1 不同浓度级别下依帕司他对照品溶液的线性关系
1.2.3 取样回收率供试品溶液的制备
根据与产品直接接触的设备、容器、工器具的材质,选择316L 不锈钢、聚四氟乙烯、有机玻璃和硅胶四种材质进行取样回收率测试。
316L 不锈钢板:精密量取对照品贮备液1 mL 均匀地滴加在316L 不锈钢板的方块区域上,方块为10 cm×10 cm,常温晾置约20 min,取布签,用乙醇约0.2 mL 润湿,将其按在涂好并晾置后的不锈钢板上,尽量使布签与取样点表面完全接触,平稳而缓慢地按图1(a) 所示方向均匀擦拭一遍,然后翻转布签,用另一面按图1(b) 所示方向进行第二次擦拭。同法重复3 次。取样必须尽快完成以免溶剂挥发和污染物沉积到样品表面。将在同一块平板上取样的布签置于同一顶空瓶中,加入10 mL 流动相,超声5 min,摇匀,过滤,作为取样回收率供试品溶液。

图1 布签擦拭示意图
聚四氟乙烯板、有机玻璃板、硅胶板的相关供试品溶液制备过程同316L 不锈钢板。
2 方法和结果
2.1 色谱条件的选择和优化
以《日本药典》[7]中依帕司他含量测定的检测方法为参考,根据紫外分光光度计检测结果,对检测波长进行调整,确定以294 nm 作为检测波长。在波长294 nm 下,考察了不同流动相比例对出峰时间和峰型的影响,发现原定流动相比例条件下布签空白溶液在依帕司他主成分峰位置出现干扰峰。后调整流动相为磷酸盐缓冲液-乙腈(65∶35),此时无明显杂峰干扰,且出峰时间有所提高。在检测波长和流动相比例同时优化的条件下,将主成分峰出峰时间提高近一倍,大大缩短了检验周期。
2.2 专属性
取适量乙醇作为空白溶剂溶液。取布签,用乙醇约0.2 mL 润湿,将其分别按在试验用的空白316L 不锈钢板(10 cm×10 cm)、聚四氟乙烯板(10 cm×10 cm)、有机玻璃板(10 cm×10 cm)、硅胶板(10 cm×10 cm) 上,按1.2.3 所述方法制备,即为布签空白溶液。
取空白溶剂溶液、布签空白溶液、对照品溶液适量,用1.2.1项下色谱条件进行色谱分析,记录各色谱图。结果表明,空白溶剂、空白布签及空白材质对依帕司他残留物含量的测定均不产生干扰,供试品溶液中主峰与相邻峰的分离度为2.45,分离情况良好。
2.3 溶液稳定性
取4℃下放置的对照品溶液和供试品溶液,分别于0、6、12、18、24、30、36、42、48 h分别进样采集,记录色谱图,结果显示,4 ℃下放置的对照品溶液及供试品溶液在48 h 内稳定,其各时间点的保留时间与初始测得值的比值在0.98~1.02 之间,各个时间点测得的主成分峰面积与初始峰面积的相对平均偏差均小于1.0%。
2.4 检测限与定量限
以信噪比约为3∶1 时的浓度作为该方法的检测限,以信噪比不小于10 时的浓度作为该方法的定量限。结果显示,该方法的检测限浓度为0.000 766 2 μg/mL,限度为0.000 076 62 μg/cm2;定量限浓度为0.003 065 μg/mL,限度为0.000 306 5 μg/cm2。
2.5 重复性
取依帕司他对照品溶液适量,按1.2.1 项下色谱条件进行色谱分析,平行进样6 次,记录其色谱图中主成分的峰面积,计算出其RSD。结果表明,6 次测得主成分峰面积RSD 为0.1%,精密度较好,符合测定的要求。
2.6 取样回收率
按1.2.3 项下配制取样回收率供试品溶液,各材质分别由两名人员各擦拭3 次,得到每种材质6 份供试品溶液。以外标法进行定量分析,结果如表2 所示。结果表明,取样回收率均大于70%,RSD 值小于20%,符合制剂生产中清洁到位的要求。

表2 各材质取样回收率结果
3 结语
建立了高效液相色谱法测定制剂生产清洁过程中依帕司他片的主成分残留量,通过优化色谱条件,大大缩短了检测周期和时间成本。对该方法专属性、线性、稳定性、检测限与定量限、精密度和回收率进行验证,结果表明该方法满足《药品生产验证指南》[8]相关要求,适用于依帕司他片清洁验证的残留物检测,为大生产过程中依帕司他片清洁验证提供了一种准确、快速且可靠的残留物检测方法。
