多价环状模板法合成交联胶束和类环胶体
2021-11-15彭金磊刘芳君
张 淼,彭金磊,刘 映,刘芳君,马 伟,魏 华
(1.南华大学药学院,湖南省分子靶向新药协同创新中心,衡阳421001;2.兰州大学化学化工学院,应用有机化学国家重点实验室,甘肃省有色金属化学与资源利用重点实验室,兰州730000)
环状结构材料(包括环状聚合物和环状胶体)由于具有丰富的拓扑结构和不同于线性类似物的独特的物理化学性质而备受关注[1,2].在相同的分子量(Mw)下,环状聚合物的玻璃化转变温度(Tg)更高,特性黏度更低,并且热稳定性更高[3~8].对于药物控释应用而言,此类结构独特的特性使其性能优于其线性类似物,包括增强的胶体稳定性、更高的细胞吸收效率和对癌细胞更高的体外细胞毒性[9~16].目前,通过扩环聚合和闭环反应合成环状聚合物的方法都存在固有的局限性.扩环法通常需要特定的单体结构和独特的催化剂,而闭环反应则需要复杂的化学合成过程和高纯度要求[17~21].因此,寻找新的环状聚合物合成方法已成为亟待解决的问题.
环糊精是一种具有独特分子结构的天然环状低聚糖分子,其自发现以来就一直备受关注[22,23].环糊精的亲水性外缘和内部独特的疏水空腔可以与客体分子络合[24,25],并且可以通过主客体识别作用构建基于环糊精的不同拓扑结构,如线性、环状、星形和网状结构[26~28].最近报道的以三臂星状聚合物作为刻蚀模板,精确合成环状胶体和环状聚合物的研究已被证实是对目前广泛采用的环状聚合物的闭环反应和扩环聚合(REP)的有效补充[29].但该模板在极稀浓度(0.3 mg/mL)的稀释条件下才会形成作为环状结构前体的单分子胶束.因此,开发一种以更高的浓度生产交联胶束和类环胶体的模板具有重要意义.
本文通过使用一种易于制备且性能良好的环状聚合物——环状聚甲基丙烯酸羟乙酯[c-P(HEMA)50]作为多价模板可控合成了交联胶束和类环胶体.首先通过点击偶联法,用二茂铁(Fc)修饰c-P(HEMA)50的侧链羟基,得到二茂铁修饰的多价环状聚合物模板c-P(HEMA-Fc)50,该模板可通过β-CD/Fc的主客体识别作用,在Fc位点有序偶联以亲水聚乙二醇(PEG)和硫辛酸(LA)功能化的β-环糊精(β-CD),形成具有明显核壳结构的超分子胶束;所得到的超分子胶束可进一步通过加入催化剂量的二硫苏糖醇(DTT)引发β-CD上修饰的LA单元的分子内的自交联反应,得到交联的超分子胶束,该交联超分子胶束可以在1.0 mg/mL的N,N-二甲基甲酰胺(DMF)和水溶液中稳定存在,以用于大规模制备交联胶束和类环胶体.进一步研究了交联超分子胶束的自组装行为和体外抗癌药物传递性能.与三臂星状聚合物模板相比,环状聚合物为模板的优势在于环状多价结构具有更高的稳定性和空间位阻效应,以其为模板制备的交联胶束和类环胶体的胶束前驱溶液的浓度可显著提高至1.0 mg/mL.利用环状多价模板制备环状胶体的过程如Scheme 1所示.
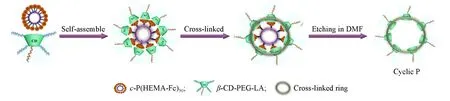
Scheme 1 Schematic illustration of a cyclic template with multivalency for the production of ring⁃like colloids
1 实验部分
1.1 试剂与仪器
2-溴代异丁酰溴(iBuBr,98%)、2,2′-联吡啶(bpy,98.0%)、4-二甲氨基吡啶(DMAP,99%)和三乙胺(TEA,99%),分析纯,北京百灵威科技有限公司;二茂铁甲酸(Fc-COOH)、DL-硫辛酸(LA)、二环己基碳二亚胺(DCC,99%)、N,N,N’,N’,N”-五甲基二亚乙基三胺(PMDETA,99%)和对甲苯磺酰氯(p-TsCl,99%),分析纯,上海阿拉丁生化科技股份有限公司;N,N-碳酰二咪唑(CDI)、聚乙二醇单甲醚(mPEG350)、二硫苏糖醇(DTT)和溴化亚铜(CuBr,99.999%),分析纯,西格玛奥德里奇(上海)贸易有限公司;β-环糊精(β-CD,96%,Mr=1134.98),分析纯,梯希爱(TCI)(上海)化成工业发展有限公司,使用前需真空干燥除水;甲基丙烯酸羟乙酯(HEMA,99%),分析纯,上海阿拉丁生化科技股份有限公司,使用前需要通过碱性Al2O3色谱柱除去阻聚剂;叠氮化钠(NaN3),分析纯,上海三友试剂厂;无水乙醚和丙酮,分析纯,衡阳市凯信化工试剂股份有限公司;2-异丙醇(IPA,99.0%)、N,N-二甲基甲酰胺(DMF)、四氢呋喃(THF)、二氯甲烷(DCM)、乙酸乙酯和正己烷,分析纯,利安隆博华(天津)医药化学有限公司,直接使用;无水二氯甲烷(DCM)使用前需在N2气回流保护下重新蒸馏;透析袋(截留分子量为3500,1000和10000),西安优博生物科技有限公司;胎牛血清和达尔伯克改良伊格尔培养基(DMEM),美国Gibico公司.
JEOL-ECS型核磁共振波谱仪(400 MHz,NMR),日本JEOL公司,以CDCl3和DMSO-d6作为氘代试剂;NEXUS 670型傅里叶变换红外光谱仪(FTIR),美国Nicolet公司,溴化钾压片法,测试范围4000~400 cm−1;DAWN EOS型体积排除色谱与多角度激光光散射联用仪(SEC-MALLS),美国Agilent公司,使用HPLC级含0.1%(质量分数)溴化锂的DMF为洗脱剂,测试流速1.0 mL/min,柱温60℃;Zetasizer Nano ZS型动态激光光散射仪(DLS),英国Malvern公司;H-7000型透射电子显微镜(TEM),日本日立公司,测试时加速电压为100 kV;Nanite型原子力显微镜(AFM),瑞士Nanosurf公司;Multifuge XIR型高速离心机,美国Thermo Scientific公司;Zetasizer Nano ZS型动态激光光散射(DLS),英国Malvern公司.
1.2 实验过程
1.2.1 小分子引发剂2-溴异丁酸丙炔酯(Alkyne-Br)的制备 将重新蒸馏后的2.0 g丙炔醇和6.8 g三乙胺加入装有50 mL无水DCM的150 mL圆底烧瓶中,冰浴搅拌30 min;将用无水DCM溶解的9.0 g 2-溴代异丁酰溴溶液缓慢滴入上述混合溶液中,待反应液滴加完成后继续冰浴搅拌1 h,然后将烧瓶转移到室温下,继续搅拌反应12 h.反应完成后对溶液进行抽滤,收集滤液,并用蒸馏水洗涤4次,溶液用无水硫酸钠进行充分干燥,旋转蒸发除去DCM,以体积比为5/1的乙酸乙酯和正己烷溶液为洗脱剂柱层析粗产品,旋转蒸发除去有机溶剂,真空干燥,得到无色油状产物2-溴异丁酸丙炔酯,产率80%.图S1(见本文支持信息)为2-溴异丁酸丙炔酯的1H NMR图.
1.2.2 二茂铁甲酸丙炔酯的制备 将2.30 g(10.00 mmol)二茂铁甲酸和2.44 g(20.00 mmol)DMAP用30 mL无水DCM充分溶解后,转移到圆底烧瓶中于冰浴下搅拌;将溶有3.10 g(15.00 mmol)DCC和1.24 g(20.00 mmol)乙二醇的10 mL DCM溶液在冰浴下缓慢滴入上述圆底烧瓶中进行反应,滴加完成后在室温下继续反应12 h;反应完成后抽滤,将滤液浓缩后以硅胶柱纯化产物,真空干燥后得到二茂铁甲酸丙炔酯橙红色固体,产率65%.图S2(见本文支持信息)为二茂铁甲酸丙炔酯的1H NMR谱图.
1.2.3 l-P(HEMA)50-Br的制备 将0.236 mmol(48.38 mg)小分子引发剂Alkyne-Br,11.8 mmol(1.44 mL)HEMA和0.471 mmol(73.10 mg)2,2′-联吡啶(bpy)溶于1.458 mL DMF和0.195 mL IPA的混合溶液中,待完全溶解后,将混合溶液转移至25 mL聚合反应管中,拧紧聚合管,进行3次冷冻-抽气-解冻循环后,在N2气保护下,将0.236 mmol(33.98 mg)CuBr迅速用纸筒加入到聚合反应管底部,继续进行3次冷冻-抽气-解冻循环,封管,于65℃搅拌反应30 min后,打开聚合管,使其与空气接触淬灭反应,并加入2 mL DMF稀释黏稠的深棕色反应物;将该反应物滴加至过量的冰乙醚中沉淀出粗产物,将粗产物用少量DMF溶液溶解,并转移至截留分子量为3500的透析袋中,用水透析48 h,每24 h换水1次,以保证产物中没有残留的铜催化剂和未反应的单体,最后冷冻干燥透析液,得到白色固体l-P(HEMA)50-Br,产率87%.
1.2.4 线性前体l-P(HEMA)50-N3的制备 在装有磁子的25 mL圆底烧瓶中分别加入800 mg l-P(HEMA)50-Br、154.94 mg NaN3和10 mL DMF,于45℃搅拌反应48 h;反应结束后,将反应液直接转移到截留分子量为3500的透析袋中,用蒸馏水透析48 h,每12 h换一次水,以去除残留的钠盐;冷冻干燥,得到白色固体l-P(HEMA)50-N3,产率90%.
1.2.5 环状聚合物c-P(HEMA)50的制备 在装有磁子的1000 mL三颈瓶中加入450 mL DMF,接入冷凝回流装置,缓慢升温至100℃,向DMF中持续通入高纯N2气鼓泡除氧1 h,然后向三颈瓶中依次加入187.68μL PMDETA和128.94 mg CuBr;同时,用10 mL DMF溶解300 mg l-P(HEMA)50-N3,小流速通入N2气鼓泡1 h以尽可能除去溶液中的氧气;将除去氧气的l-P(HEMA)50-N3溶液以0.4 mL/h的速度用注射泵缓慢注入CuBr/PMDETA催化体系中,注射完成后于100℃反应24 h;反应结束后,待反应液冷却至室温后,于80℃旋转蒸发除去大量的DMF;然后将剩余的溶液(5 mL左右的DMF溶液)转移到截留分子量为3500的透析袋中用水透析;透析24 h后,离心除去铜催化剂,将上清液继续用水透析36 h;冷冻干燥得到白色絮状产物c-P(HEMA)50,产率67%.
1.2.6 c-P(HEMA-Br)50大分子引发剂的制备 将100 mg c-P(HEMA)50溶解在4 mL无水DMF中,在冰水浴中搅拌,并持续通入N2气;在反应混合物中逐滴加入324μL 2-溴代异丁酰溴,滴加完成后继续于0℃下搅拌1 h,然后在室温下搅拌24 h;待反应结束后用过量的冰乙醚对产物进行沉淀,得到的固体沉淀物用2 mL DMF溶解,并转移到截留分子量为3500的透析袋中用水透析48 h,每24 h换水1次;将透析液冷冻干燥,得到白色固体粉末c-P(HEMA-Br)50,产率95.3%.
1.2.7 环状聚合物c-P(HEMA-N3)50的制备 将135.28 mg(0.0045 mmol)c-P(HEMA-Br)50与311.39 mg(10.06 mmol)NaN3溶于3 mL无水DMF溶液中,于45℃搅拌反应48 h;反应结束后,将混合反应液直接转移到截留分子量为3500的透析袋中用水透析48 h;冷冻干燥,得到淡黄色固体粉末c-P(HEMAN3)50,产率88.7%.
1.2.8 环状聚合物模板c-P(HEMA-Fc)50的制备 将20 mg c-P(HEMA-N3)50和28.51 mg二茂铁甲酸丙炔酯溶于3 mL DMF溶液中,加入17μL配体PMDETA形成均相;将混合溶液转移至25 mL聚合反应管中,封管,进行3次冷冻-抽气-解冻循环,打开N2气袋,用纸筒迅速将11.74 mg CuBr催化剂加入到聚合管底部,重复上述冷冻-抽气-解冻循环3次;封管,在50℃油浴锅中反应24 h,反应结束后,先用冰乙醚进行沉淀,离心,粗产物用THF溶解后过中性Al2O3柱子,去除未反应完的铜催化剂,真空干燥,得到片状黄褐色固体c-P(HEMA-Fc)50,产率89.6%.Scheme 2为c-P(HEMA-Fc)50的合成示意图.
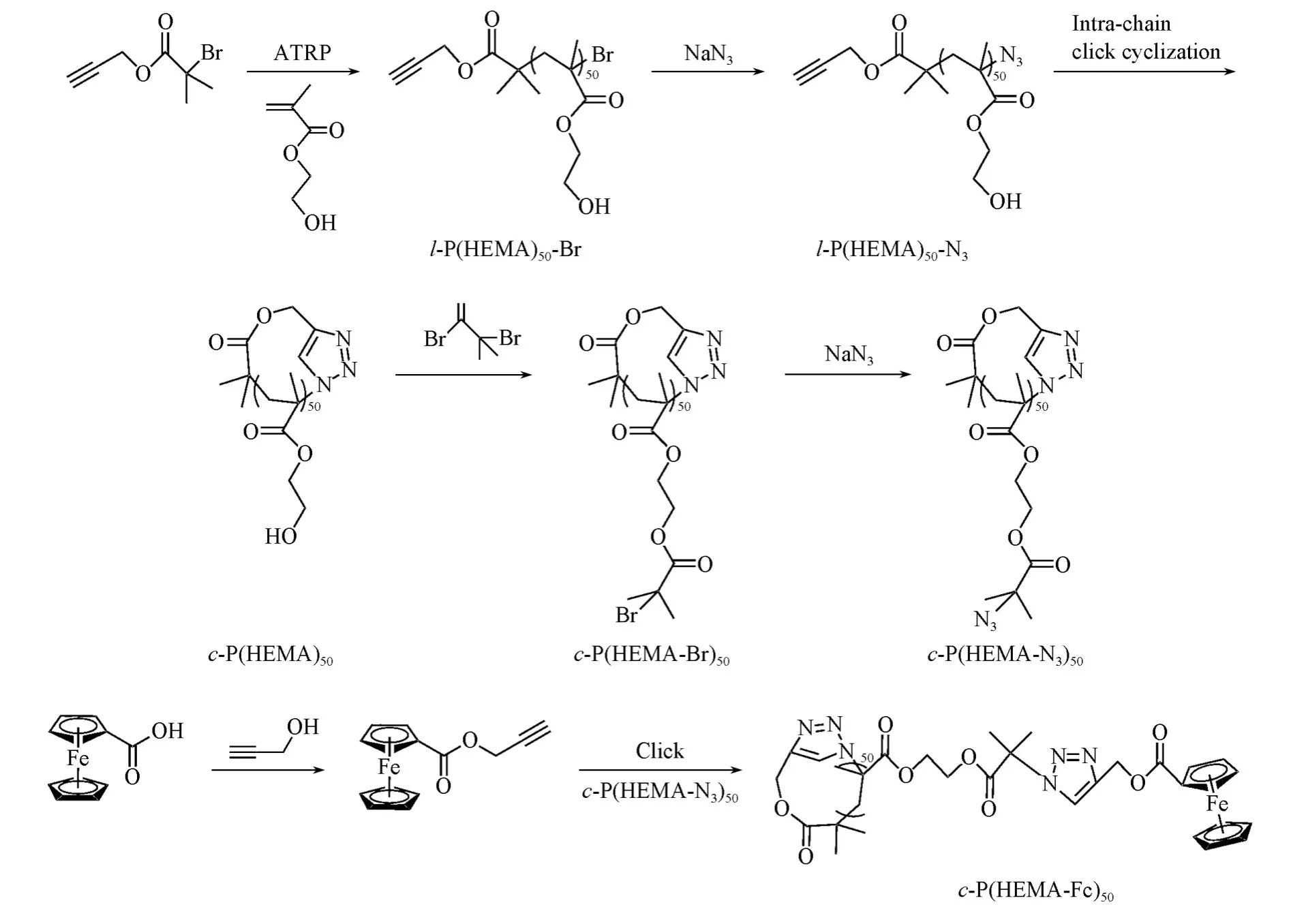
Scheme 2 Synthesis of c⁃P(HEMA⁃Fc)50
1.2.9β-CD-(mPEG350)5的制备 将2.37 gN,N-碳酰二咪唑(CDI)和2.56 g mPEG350溶于20 mL无水DCM中;将上述溶液转移至50 mL圆底烧瓶中反应24 h;补加10 mL无水DCM,用30 mL饱和NaCl溶液洗涤5次,每次从下层油相中收集产物,加入无水Na2SO4干燥4~5 h,过滤后旋转蒸发至干,用油泵将产物中残留的DCM溶液抽干,将抽干的产物放入冻干机中继续干燥,得到淡黄色黏稠状液体CDI-(mPEG350)5,产率48%.
将639.36 mg CDI-(mPEG350)5用5 mL无水DMF溶解,加入0.5 mL TEA,转移至25 mL圆底烧瓶中;再用2.5 mL无水DMF溶解272.4 mg干燥的CD,转移至恒压滴液漏斗中,缓慢滴加入圆底烧瓶中,常温搅拌反应24 h;反应结束后,反应液用无水乙醚沉淀2次,冷冻干燥,得到黏而硬的白色透明产物β-CD-(mPEG350)5,产率73.6%.
1.2.10β-CD-(mPEG350)5-LA6的制备 将215.22 mgβ-CD-(mPEG350)5、88.37 mg DCC和43.60 mg DMAP溶于4 mL无水DCM中;将溶液转移至25 mL聚合反应管中,拧紧聚合管,进行冷冻-抽气-解冻循环3次,在N2气保护下,于黑暗中将73.64 mg硫辛酸迅速加入聚合管底部;重复上述冷冻-抽气-解冻循环3次,封管,避光冰水浴搅拌1 h后撤去冰浴,室温避光搅拌24 h;反应完成后过滤除去生成的N,N-二氯氨基甲酸乙酯(DCU)副产物,用冰乙醚沉淀2次,离心,冷冻干燥后得到黄色黏稠状键合硫辛酸的亲水段β-CD-(mPEG350)5-LA6,产率68.4%.Scheme 3为β-CD-(mPEG350)5-LA6的合成示意图.
1.2.11 超分子胶束[c-P(HEMA-Fc)50/β-CD-(mPEG350)5-LA6]的制备及交联 用1 mL DMF溶解21.36 mg疏水段聚合物c-P(HEMA-Fc)50,然后转移到2.64 mgβ-CD-(mPEG350)5-LA6亲水段的离心管中,使其充分溶解后,通N2气5 min,室温下避光超声1.5 h;将超声后的聚合物溶液用注射泵以0.5 mL/h的速度缓慢注入到24 mL通N2气的去离子水中,注射完成后再搅拌2 h,以确保成功形成超分子胶束;搅拌完成后,将上述聚合物水溶液直接转移到截留分子量为10000的透析袋中,用去离子水透析24 h以除去有机溶剂DMF,将透析所得聚合物水溶液冷冻干燥,得到c-P(HEMA-Fc)50/β-CD-(mPEG350)5-LA6自组装体.
用1 mL DMF溶解21.36 mgc-P(HEMA-Fc)50疏水段聚合物,转移到装有2.64 mgβ-CD-(mPEG350)5-LA6亲水段的离心管中,待其充分溶解后,通入N2气5 min,室温下避光超声1.5 h;将超声后的聚合物溶液用注射泵以0.5 mL/h的速度缓慢注入到10 mL通N2气的去离子水中,注射完成后再搅拌2 h;用截留分子量为10000的透析袋用水透析2 h;补入14 mL去离子水,转移至50 mL圆底烧瓶中,通N2气搅拌1 h;将2.03 mg催化剂DTT(硫辛酸摩尔数的10%)用1 mL超纯水溶解,用移液枪取33.84μL DTT溶液加入到上述24 mL自组装溶液中,避光搅拌24 h;反应结束后,直接转移到截留分子量为10000的透析袋中用水透析24 h,冷冻干燥,得到交联产物c-P(HEMA-Fc)50/β-CD-(mPEG350)5-LA6(CLP).
1.2.12 环状核模板的刻蚀 将交联后的3 mg聚合物溶于2 mL DMF溶液中,转移至截留分子量为100000的透析袋中,于含有500 mL DMF的1 L烧杯中透析,加热至50℃,以脱除环状疏水聚合物模板,透析 72 h,每24 h更换一次DMF;有机溶剂透析结束后,将透析袋转移至5 L去离子水中透析24 h;将透析溶液冷冻干燥,得到类环聚合物(Cyclic P),产率71.4%.
1.2.13 交联载药胶束的体外细胞毒性 将包药后的聚合物溶于水,配成母液,逐渐稀释为一系列浓度的聚合物样品,将其接种在OptiMEM培养基上;将细胞铺在细胞密度为2500 Cell/孔的96孔板上,在37℃、5%CO2的培养基中培养24 h,然后用PBS缓冲溶液洗涤细胞;将40μL预先接种好的样品加入到上述培养基中,在37℃下继续培养4 h;培养后的细胞用PBS缓冲溶液洗涤,并加入新鲜的培养液,继续培养24 h;在每个孔板上加入20μL MTS试剂,在上述培养箱中继续培养细胞3 h;通过Tecan Safire 2平板读数器测定每个孔在490 nm处的吸光度值,计算出加入上述材料后细胞的存活率.

Scheme 3 Synthesis ofβ⁃CD⁃(mPEG)5⁃LA6
2 结果与讨论
2.1 环状聚合物模板c-P(HEMA-Fc)50的制备与表征
采用ATRP与点击化学相结合的方法合成目标聚合物c-P(HEMA-Fc)50.主要合成步骤如下:(i)采用ATRP技术,以Alkyne-Br引发单体HEMA聚合,制备线性聚合物l-P(HEMA)50-Br;(ii)通过线性聚合物l-P(HEMA)50-Br末端溴与叠氮基进行亲核取代反应制备线性前体l-P(HEMA)50-N3;(iii)在极稀条件下,通过分子内点击环化线性前体l-P(HEMA)50-N3制备环状聚合物c-P(HEMA)50;(iv)环状聚合物c-P(HEMA)50与过量的iBuBr进行酯化反应,得到环状大分子引发剂c-P(HEMA-Br)50;(v)环状大分子引发剂c-P(HEMA-Br)50与NaN3反应,得到末端含有叠氮基的环状聚合物c-P(HEMA-N3)50;(vi)c-P(HEMA-N3)50与二茂铁甲酸丙炔酯进行点击耦合反应,制备二茂铁修饰的环状聚合物模板c-P(HEMA-Fc)50.
首先通过ATRP技术合成了线性聚合物l-P(HEMA)50-Br,其1H NMR谱图见图S3(见本文支持信息)上出现明显的HEMA单元的特征信号峰,根据δ4.60处与炔基相连的亚甲基质子b峰和PHEMA与羰基相连的亚甲基质子f峰及其侧链末端的羟基质子h峰的积分强度比,得到HEMA单元的聚合度约为50.同时图S4(见本文支持信息)显示l-P(HEMA)50-Br在DMF溶液中的SEC-MALLS淋洗曲线呈单峰窄分布.通过线性末端溴与NaN3进行亲核取代反应,使l-P(HEMA)50-Br末端带上可点击的叠氮基.叠氮化后,SEC-MALLS分析表明,其SEC-MALLS淋洗曲线呈单峰分布,且整体稍微向右移动,表明成功进行了叠氮化反应(见本文支持信息图S4).
在极稀的DMF溶液中链内点击环化线性前体l-P(HEMA)50-N3,成功制备了目标聚合物环状c-P(HEMA)50.图S5(见本文支持信息)中δ4.60处烷基乙烯的乙烯质子峰消失;与线性聚合物相比,环状聚合物由于缺少链末端而流体力学体积较小,因此,其SEC淋洗曲线整体向右偏移(低分子量)(图S4);在FTIR谱图(图S6,见本文支持信息)中,2100 cm−1处的叠氮基特征吸收峰消失.以上结果均证实成功进行环化反应,合成了环状聚合物c-P(HEMA)50.
通过酯化反应制备了环状大分子引发剂c-P(HEMA-Br)50.通过比较图S5中c-P(HEMA)50和图S7(见本文支持信息)中c-P(HEMA-Br)50的1H NMR谱图可以发现,在图S7中图S5中δ4.79处HEMA单元的末端羟基峰消失了,同时在δ1.90处出现了新的信号较强的质子峰a,这是发生酯化反应后紧挨着Br的甲基的质子峰,由此证明c-P(HEMA)50上的羟基被Br完全取代.
c-P(HEMA-Br)50的末端溴与NaN3发生亲核取代反应,叠氮化后,SEC-MALLS分析表明,其SECMALLS淋洗曲线呈单峰分布,且其SECMALLS曲线整体稍微向右移动,表明成功进行了叠氮化反应(图S8,见本文支持信息).将c-P(HEMA-N3)50与二茂铁甲酸丙炔酯进行分子间点击反应制备侧链含有二茂铁单元的环状聚合物模板c-P(HEMA-Fc)50.图1给出c-P(HEMA-Fc)50的1H NMR谱图,其中的质子峰a,b和c为二茂铁单元的特征质子峰,通过计算δ1.75处的j峰与二茂铁在δ4.3处的质子b峰和δ5.25处的质子c峰的积分强度比,得到点击效率接近100%,并且图S8中SEC-MALLS淋洗曲线明显前移,表明点击反应是成功的.

Fig.1 1H NMR spectrum of c⁃P(HEMA⁃Fc)50 in DMSO⁃d6
2.2 亲水段β-CD-(mPEG350)5-LA6的制备与表征
通过CDI与单体mPEG350进行酯化反应得到β-CD-(mPEG350)5;再进一步通过DCC缩合反应,利用环糊精多羟基位点与硫辛酸(LA)功能化的环糊精制备β-CD-(mPEG350)5-LA6.
通过酯化反应得到CDI-(mPEG350)5,其1H NMR谱如图S9(见本文支持信息)所示,由δ3.3处mPEG350链末端的甲基质子a峰及与CDI相连的酯基旁边的亚甲基质子b峰积分面积得到mPEG350的聚合度为5,同时在δ8.2,7.5和7.2处分别出现CDI的特征氢峰(e,c和d峰).
通过环糊精与硫辛酸进行DCC缩合反应制备亲水段,其1H NMR谱如图2所示,根据mPEG350链末端的甲基质子a峰与硫辛酸的特征氢b,c和d峰的积分面积比,计算得到硫辛酸的键合数为6.图3给出了所制备的两亲性聚合物[c-P(HEMA-Fc)50/β-CD-(mPEG350)5-LA6]的SEC-MALLS淋洗曲线.所有制备的聚合物的分子参数列于表1.

Fig.2 1H NMR spectrum ofβ⁃CD⁃(mPEG)5⁃LA6 in CDCl3

Fig.3 SEC⁃MALLS elution traces ofβ⁃CD⁃(mPEG)5⁃LA6,c⁃P(HEMA⁃Fc)50,cross linking P(CLP)and cyclic P using DMF as an eluent
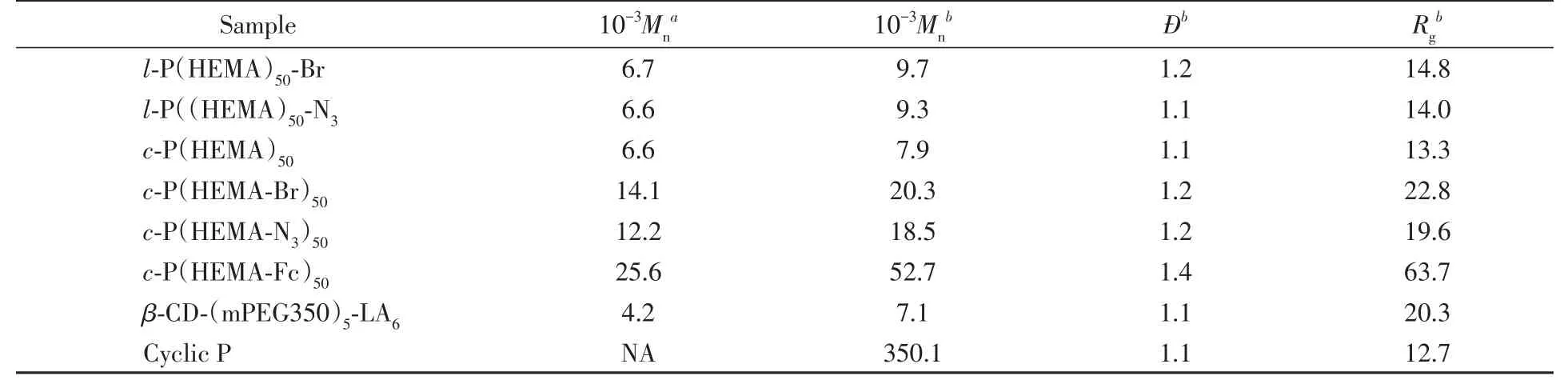
Table 1 Summary of molecular parameters of all the synthesized polymers

Fig.4 2D NOESY NMR spectra of c⁃P(HEMA⁃Fc)50 andβ⁃CD⁃(mPEG350)5 in DMSO⁃d6
2.3 超分子胶束的制备与表征
将亲疏水段聚合物溶于有机溶剂DMF中,超声30 min后将其缓慢注射到大量超纯水中,得到均相且澄清透明的聚合物溶液.图4给出β-CD-(mPEG350)5和c-P(HEMA-Fc)50的二维NOESY谱图,证明成功制备了β-CD/Fc主-客体包合物.紫色圆圈中突出的交叉峰归属于β-CD质子在δ3.25~3.60范围内的特征吸收峰与Fc在δ4.20~4.50范围内的共振吸收峰之间的偶极相互作用.环状核模板的多价结构使所制备的超分子核-壳胶束比先前报道的基于三臂星形聚合物模板的胶束前体具有更高的稳定性和空间位阻,从而显著提高了制备交联胶束和类环胶体的浓度(1.0 mg/mL,先前使用的环状胶体的浓度为0.3 mg/mL).在37℃,聚合物浓度为1.0 mg/mL时通过动态光散射(DLS)测得c-P(HEMA-Fc)50与β-CD-(mPEG350)5-LA6自组装胶束的平均流体动力学直径为142.6 nm(图5).根据SEC-MALLS测 定 的c-P(HEMA-Fc)50(63.7 nm)和β-CD-(mPEG350)5-LA6(20.3 nm)的Rg值(表1),超分子胶束的直径约为100 nm,与DLS数据吻合较好.该超分子胶束具有优异的稳定性,其在0.03~1.0 mg/mL的各种稀释浓度几乎保持相同的流体力学尺寸和尺寸分布,这归因于环状聚合物作为模板时可能处于具有更大稳定性和空间位阻的多价结构中,从而导致制备环状材料的胶体浓度显著增加.同时,该胶束在水相和DMF相的流体力学体积表现出几乎相同的尺寸及尺寸分布[图6(A)].以上结果证实,环状聚合物作为多价核心模板具有更大的稳定性和空间位阻性,有利于环类胶体和环状聚合物的大规模生产.

Fig.5 Mean hydrodynamic size and size distribution of P micelles at various concentrations ranging from 0.03 mg/mL to 1.2 mg/mL

Fig.6 Mean hydrodynamic size and size distribution of P micelles in water and DMF at a polymer concentration of 1.0 mg/mL(A)and NCLP,CLP and Cyclic P micelles in a polymer concen⁃tration of 1.0 mg/mL(B)
2.4 DTT催化的交联胶束的制备与表征
向c-P(HEMA-Fc)50/β-CD-PEG-LA的超分子胶束中进一步加入催化剂量的二硫苏糖醇(DTT),引发β-CD上修饰的LA单元的分子内的自交联反应,得到交联的超分子胶束(CLP),作为下一步环状结构的刻蚀模板.临界胶束浓度对于制备CL胶束前体至关重要,一方面,该浓度应足够低,以确保仅发生胶束内交联而没有任何胶束间反应的发生;另一方面,该浓度也应该足够高,以允许以适当的产率大规模生产环状胶体.由于使用的环状核模板具有多价性、稳定性和增强的空间位阻,本文制备环状胶体的前体浓度(1.0 mg/mL)明显高于以三臂星状聚合物作为模板时的浓度(0.3 mg/mL).图6(B)中未交联超分子胶束(NCLP)和交联超分子胶束几乎一致的流体力学直径表明在此关键制剂浓度下的胶束内交联占主导地位.利用SEC-MALLS进行脱核后的表征,选择DMF为洗脱剂,柱子的温度为60℃.可以发现,交联后SEC洗脱曲线明显前移,并且出现两个SEC洗脱峰,其保留时间分别为16 min及20 min左右(图3).根据先前的文献报道,DMF是亲疏水段的良溶剂,当在较高温度(50℃)时,环糊精会与客体分子之间发生解组装[29],因此出现的双峰中具有较短保留时间的主峰就合理地归属为亲水段交联后脱去环状模板c-P(HEMA-Fc)50的洗脱峰,并且其保留时间与c-P(HEMA-Fc)50相符合.另外环状聚合物的SEC-MALLS洗脱峰向较短的洗脱时间明显转移,通过计算,环状P的Mn(Mn=350100)约是单个亲水段β-CD-(mPEG350)5-LA6(Mn=7100)的50倍(表1),这一结果证明通过基于P胶束的充分交联成功地形成了包含50个β-CD-(mPEG350)5-LA6亲水段的环状胶体.上述结果证实了使用DMF和相对较高的温度来刻蚀环状核模板c-P(HEMA-Fc)50的可行性.
2.5 环状聚合物和类环胶体的制备与表征
在较高温度(50℃)下的DMF溶液中,通过破坏环糊精与客体分子二茂铁之间的相互作用力,刻蚀掉环状核模板c-P(HEMA-Fc)50.当胶束解组装后,通过亲疏水段聚合物分子量的差异,选择合适分子量的透析袋(100000)选择性分离c-P(HEMA-Fc)50,最后得到完全由环糊精构建的环状聚合物.脱核后SEC-MALLS淋洗曲线呈现的单峰表明疏水环状聚合物核模板已被脱除,成功制备了环状聚合物或类环胶体.通过DLS测试得到交联前c-P(HEMA-Fc)50/β-CD-(mPEG)5-LA6液体力学粒径为142.6 nm,交联后为137.1 nm,脱掉环状模板后为214.2 nm.
透射电子显微镜(TEM)测量可以直观地观察每个阶段聚合物的形态变化.未交联超分子胶束(NCLP)和交联超分子胶束(CLP)均形成大小相似且分散均匀的球形纳米颗粒,其平均直径约为35 nm[图7(A)和(B)],这也证实了交联仅发生在胶束内,而未发生任何胶束间反应.当刻蚀掉环状核模板c-P(HEMA-Fc)50后,TEM图像显示出明显的形态转变,其纳米粒子的大小已增加到大约45 nm[图7(C)].这种尺寸变化说明疏水内核的去除促使亲水部分溶胀,这与DLS结果非常吻合[图6(B)].图7(D)给出环状聚合物的AFM照片,图中清晰而均匀的环有力地证实了环状内核的刻蚀及成功生成环状聚合物.类环状聚合物的平均大小约为42 nm,这与TEM分析结果吻合.与TEM数据相比,类环状聚合物胶束在DLS下的平均尺寸略大,这可能是由于环状聚合物的状态不同,即用于TEM观测的为脱水状态而用于DLS测量的为水合状态[30].

Fig.7 TEMimages of NCLP(A),CLP(B)and Cyclic P(C)and AFMimage of Cyclic P(D)
2.6 交联载药胶束的体外细胞毒性
在人肝癌细胞HepG2中,通过MTS法评估了空白和负载抗癌药物阿霉素(DOX)的交联胶束的体外细胞毒性.由图8可见,即使在浓度高达0.48 mg/mL时,空白胶束对HepG2细胞仍表现为不抑制,细胞存活率高于80%,结果表明空白聚合物胶束对肝癌细胞基本无毒,具有良好的生物相容性.
同时,我们考察了负载DOX的交联胶束的体外细胞毒性.经GraphPad Prism 6软件计算得到自由DOX和载药交联胶束的IC50(即细胞存活率为50%对应的药物浓度)值分别为1.09(置信区间为0.87~1.32)和70.81(置信区间为60.50~81.11)μg/mL.载药交联胶束的体外细胞毒性[图9(A)]明显低于自由DOX[图9(B)],这可能是因为自由DOX具有直接的膜渗透机制,而载药交联胶束是通过较慢的内吞作用进入细胞的,同时相对于直接在胞内发挥药效的小分子DOX,载药胶束在胞内是通过药物释放动力学来实现药物的释放,说明本文制备的聚合物在药物控释与癌症治疗领域具有很大的应用潜力[31].

Fig.9 In vitro cytotoxicity of HepG2 cells incubated for 24 h with DOX⁃loaded CLP micelles(A)and free DOX(B)
3 结 论
使用一种易制备且性能良好的环状聚合物——环状聚甲基丙烯酸羟乙酯[c-P(HEMA)50]作为生产相对较高浓度的环状胶体的新型模板.该环状聚合物模板具有更高的稳定性和空间位阻的多价结构,其制备交联胶束和类环胶体胶束的前驱溶液的浓度可显著提高至1.0 mg/mL.本文不仅发展了简单高效的制备环状聚合物的新策略,并且所开发的具有聚二硫键的还原敏感性交联胶束和环状胶体在控释应用中具有巨大潜力.
支持信息见http://www.cjcu.jlu.edu.cn/CN/10.7503/cjcu20210370.
