Gli2对顺铂诱导急性肾小管上皮细胞凋亡及纤维化的影响研究*
2021-11-13赵若蓓黄爱芳潘小洁高天云
彭 珣,赵若蓓,黄爱芳,潘小洁,高天云,潘 玲
(广西医科大学第一附属医院肾内科,南宁 530021)
急性肾损伤(acute kidney disease,AKI)为临床常见的急危重症疾病,发病率高,预后不容乐观,耗费巨大的医疗资源。调查发现,发达国家AKI住院患者占住院总人数的3.2%~9.6%[1-3],AKI院内总体病死率约为10%~20%[4],国内报道显示医院住院患者AKI发病率为0.15%~11.20%,病死率为5.00%~23.33%[5-6]。AKI 不仅有高发病率及死亡率,还是慢性肾脏病(CKD)的重要原因[7]。有研究提示,高危住院患者中的AKI 约40%遗留慢性肾脏损害,10%~20%甚至需要持续性透析。目前已知的影响AKI 发生及进展的危险因素较多,相关机制复杂。其中,肾小管上皮细胞损伤及纤维化变被认为是AKI 发生进展甚至慢性化转变的关键机制[8],尤其肾小管细胞凋亡是肾纤维化重要原因,但具体机制尚不清楚。近年有研究表明,Hedgehog(Hh)/Gli 通路是肾纤维化的重要信号通路[9-10],Gli 是一种特别的血管周围间充质干细胞(MSCs)的标志物,而这种血管周围MSCs 是肌成纤维细胞的重要来源。Gli 有Gli1,Gli2 及Gli3 三种类型,其中Gli2 则是导致肾小管上皮细胞损伤及修复不良的一种锌指蛋白,也是肌成纤维细胞活化过程中的驱动因子[11],调控Hedgehog通路下游靶基因的激活和转录,引起肾小管上皮细胞凋亡,导致肾小管损伤及纤维化转变。
目前Hedgehog/Gli2 通路在AKI 发生发展中的作用及机制研究鲜少。本课题组拟运用短发夹RNA(shRNA)沉默、过表达、实时荧光定量PCR(RT-qPCR)及流式细胞仪凋亡测定等技术,通过顺铂刺激肾小管上皮细胞构建AKI 体外细胞模型进行研究,探讨Hedgehog 通路关键转录因子Gli2 在AKI 肾小管上皮细胞凋亡及纤维化转变中的作用,为AKI发生及进展的防治提供新的思路。
1 资料与方法
1.1 细胞株与主要试剂
大鼠肾小管上皮细胞NRK52E,购于中国科学院上海细胞库。DMEM 培养基购于美国Gibco 公司,顺铂购于美国Sigma公司,细胞凋亡试剂盒购自杭州联科生物公司,CCK-8试剂盒和逆转录试剂盒均购自南京诺唯赞生物科技股份有限公司,RNA提取试剂盒购自美国Axygen公司,荧光定量试剂盒购自美国ABclonal 公司。Gli2 沉默和过表达慢病毒均购自上海吉凯基因公司。
1.2 细胞培养及顺铂干预
NRK52E 细胞用含10%胎牛血清的DMEM 培养基在5%CO2、37 ℃恒温培养箱培养,待细胞汇合度达到80%~90%,胰酶消化细胞,调整细胞密度约5×105个/mL 接种至6 孔板内,分别加入0 μmol/L、12.5 μmol/L、25 μmol/L 的顺铂刺激24 h,提取总RNA,用RT-qPCR法检测Gli2表达水平的情况。
取对数生长期的NRK52E 细胞接种至6 孔板内,加入Gli2 沉默和过表达慢病毒感染48 h 后,更换新鲜培养基,加入嘌呤霉素筛选,杀死转染失败的无抗性细胞。取部分细胞,提取总RNA,用RTqPCR 法检测沉默和过表达效率。细胞分组如下:沉默空载组、沉默Gli2 组、过表达空载组及过表达Gli2 组,沉默Gli2 组感染沉默Gli2 慢病毒,沉默空载组感染沉默对照慢病毒,过表达Gli2组感染过表达Gli2慢病毒,过表达空载组感染过表达对照慢病毒,然后每组均分别用顺铂12.5 μmol/L 处理细胞24 h,建立顺铂所致肾损伤体外细胞模型。
1.3 细胞凋亡检测
顺铂12.5 μmol/L 处理各组细胞24 h 后,取105个细胞,1 200 r/min离心5 min,离心去上清,每管加入50 μL 1×Binding Buffer,重悬,按以下分组加入染料:阴性管:不加任何染料;样本管:加入5 μL Annexin-V APC 和10 μL 7-AAD,将溶液轻轻地震荡后,室温避光孵育15 min,分别加入200 μL 1×Binding Buffer,在1 h内进行流式细胞仪检测。
1.4 RT-qPCR检测肾损伤、纤维化相关指标相关表达量
细胞干预处理24 h 后收集细胞,提取细胞总RNA,逆转录成cDNA,进行RT-qPCR 检测,反应条件为:95 ℃3 min;95 ℃10 s,60 ℃1 min,40 个循环;95 ℃10 s,65 ℃1 min,97 ℃1 s。扩增引物由武汉金开瑞生物工程有限公司合成,引物序列见表1。

表1 RT-qPCR引物序列
1.5 统计学方法
采用SPSS 17.0 进行统计学分析,符合正态分布的计量资料采用均数±标准差()表示,组间比较用t检验或者方差分析(one-way ANOVA),组间两两比较采用LSD-t检验。以P<0.05 为差异有统计学意义。
2 结果
2.1 顺铂诱导肾小管上皮细胞损伤中Gli2 表达水平
随着顺铂浓度的增加,Gli2 的mRNA 表达水平不断升高(P<0.05),提示顺铂可呈剂量依赖性诱导NRK52E细胞中Gli2的表达,见图1。

图1 不同浓度的顺铂诱导NRK52E 细胞Gli2 水平表达情况(*P<0.05)
2.2 Gli2沉默和过表达效率检测
提取转染沉默Gli2 组和过表达Gli2 组慢病毒的NRK52E细胞的总RNA,分别以感染沉默和过表达慢病毒空载质粒的NRK52E 细胞为对照,结果显示沉默Gli2 组NRK52E 表达水平下调,沉默效率为65.3%,而过表达Gli2 组Gli2 表达水平上调4.56 倍(图2)。结果表明,我们成功获得了NRK52E 沉默和过表达Gli2的稳转细胞株。

图2 沉默及过表达Gli2效率检测
2.3 沉默和过表达Gli2 对顺铂处理NRK52E 细胞后AKI和纤维化指标的影响
在沉默Gli2 组和过表达Gli2 组分别予12.5 μmol/L顺铂处理NRK52E细胞24 h后,沉默Gli2组AKI 标志物NGAL 及纤维化标志物αSMA、TGFβ1的mRNA 水平低于沉默空载组,Hedgehog 通路中Smo 的mRNA 水平也显著低于沉默空载组(均P<0.05),图3;过表达Gli2组αSMA水平高于过表达空载组(P<0.05),NGAL 及TGFβ1 的mRNA 水平与对照组比较无统计学意义(均P>0.05)。
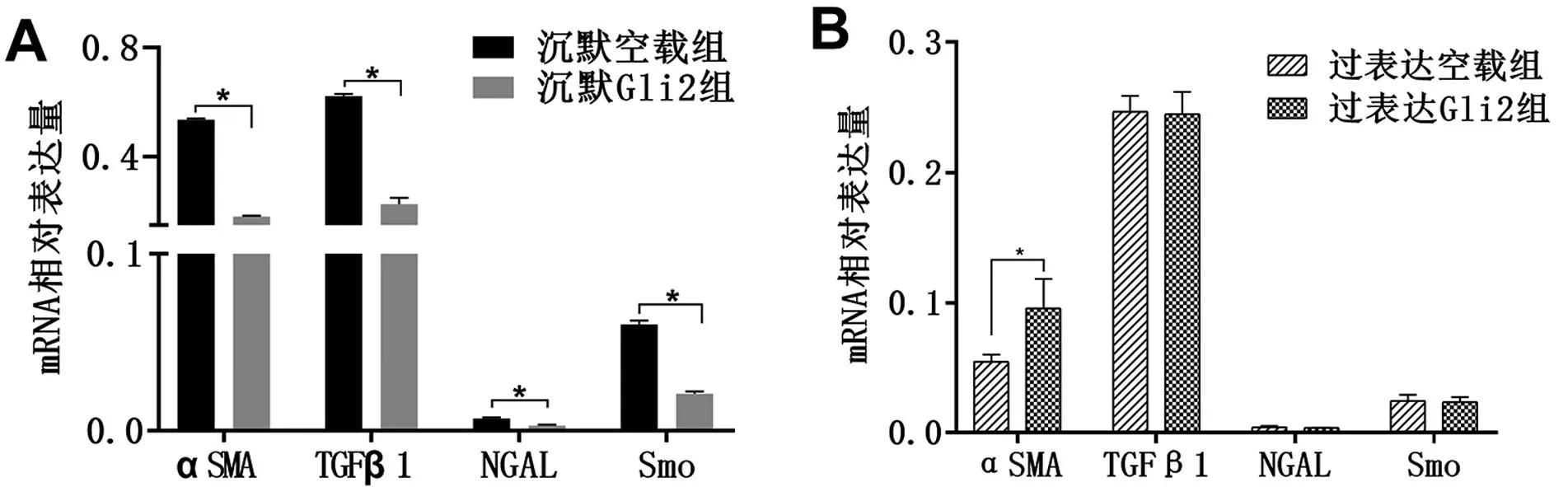
图3 沉默及过表达Gli2组顺铂刺激细胞后AKI和纤维化指标的情况
2.4 沉默和过表达Gli2 对顺铂处理NRK52E 细胞后细胞凋亡的影响
与沉默空载组比较,沉默Gli2组细胞的早期凋亡、晚期凋亡以及总凋亡百分比、促进凋亡分子Bax和凋亡相关蛋白Caspase7 的mRNA 水平均较空载组降低(均P<0.05),见图4、图6。
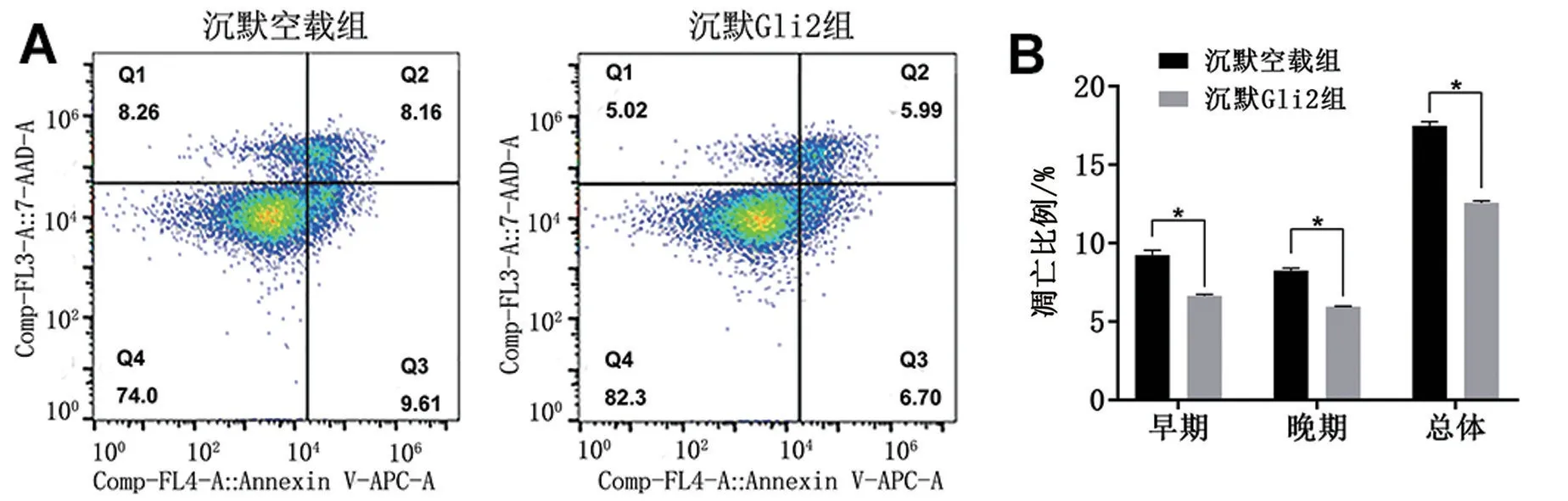
图4 沉默Gli2对顺铂致AKI的细胞凋亡的影响
与过表达空载组比较,过表达Gli2组细胞的早期凋亡、晚期凋亡以及总凋亡百分比均升高(均P<0.05),Bax、Caspase3、Caspase7 的mRNA 水平无统计学差异(均P>0.05),见图5、图6。
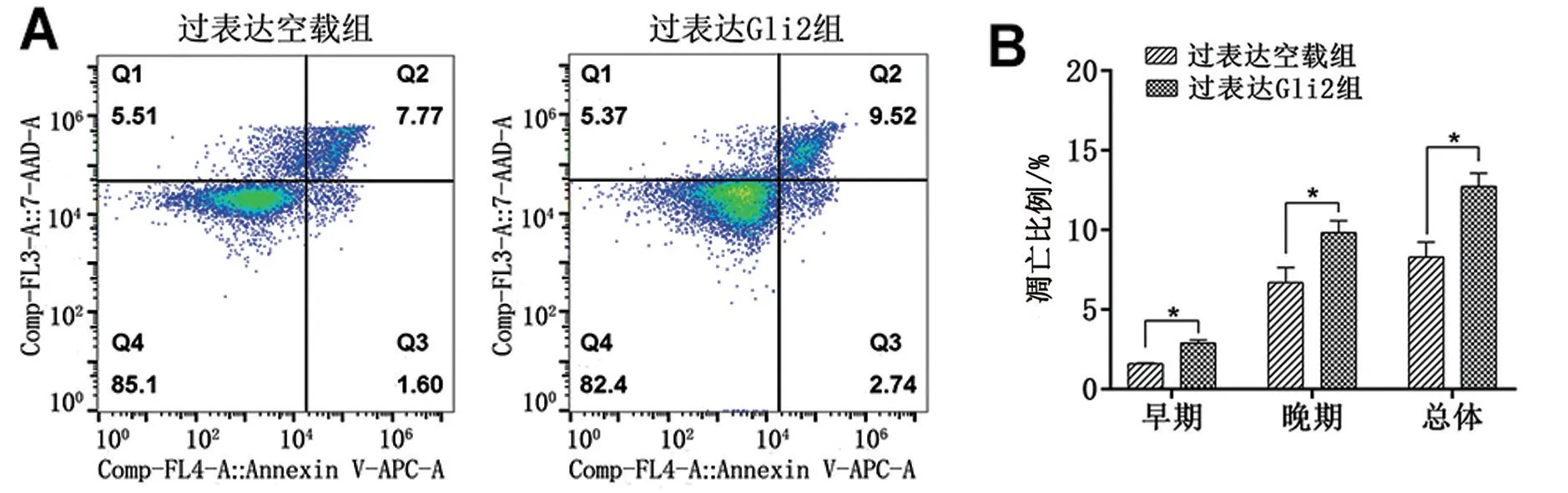
图5 过表达Gli2对顺铂致AKI的细胞凋亡的影响
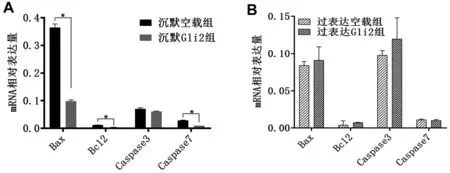
图6 沉默和过表达Gli2组顺铂刺激细胞后凋亡标志物表达的情况
3 讨论
AKI 发生及进展的机制复杂,而急性肾小管坏死(ATN)是最常见的AKI类型,目前认为ATN发生后引起肾小管上皮细胞损伤,如病因持续存在则小管上皮细胞修复不良并转化为肌成纤维细胞,如仍不能纠正则进一步纤维化转变,AKI 易发生进展至CKD 甚至慢性肾衰[12]。肾小管上皮细胞损伤是AKI 发生后慢性纤维化转变的启动机制,肾小管上皮细胞损伤是否修复不良决定AKI 的临床转归。顺铂等肾毒性化疗药物导致的AKI 是临床常见的ATN类型,顺铂诱导的ATN常用于AKI的基础实验研究模型,也被广泛用作AKI纤维化至慢性肾损伤的模型。我们的研究也是基于顺铂刺激肾小管诱导ATN 的体外细胞实验探讨Gli2 对AKI 肾小管上皮细胞凋亡和纤维化的影响。我们的研究结果提示,顺铂可诱导NRK52E 细胞中Gli2 的mRNA 表达,并呈剂量依赖性,Hedgehog 通路的关键转录因子Gli2 可影响顺铂诱导的肾小管上皮细胞凋亡及纤维化,沉默Gli2能改善顺铂介导AKI的肾小管上皮细胞凋亡,下调纤维化、AKI 标志物表达,并可下调Hedgehog 通路蛋白受体Smo 表达。而过表达Gli2 可促进肾小管上皮细胞凋亡率,上调纤维化标志物。有研究表明,顺铂可激活Hedgehog信号通路参与肾小管损伤,而这个过程中Hedgehog通路下游转录因子Gli1 及Gli2 的水平也升高,能够阻断Hedgehog 信号通路的药物可同时抑制Gli2 蛋白水平[13]。而另一横纹肌溶解后AKI 的动物试验也表明,肾小管受损情况下Hh 通路可发生滞后激活,AKI 之后的24 h 及72 h 时Hh 通路的Shh 及Gli2 表达明显上调[14],这些均提示Hedgehog/Gli 通路参与调控AKI肾小管上皮细胞损伤,而Gli2可能是引起AKI发生及进展的促进因子。此外本研究发现过表达Gli2组经顺铂刺激后,肾小管上皮细胞的早期凋亡、晚期凋亡以及总凋亡百分比均升高。而沉默Gli2组经顺铂刺激后,肾小管上皮细胞的早期凋亡、晚期凋亡以及总凋亡百分比均降低,凋亡相关的关键蛋白Bax 和Caspase7 的mRNA 水平较空载组降低。有研究也发现,Gli2水平的表达与Bcl-2表达相关,抑制Gli2表达可以抑制Bcl-2、Bax,Caspase-3和Caspase-9,加速细胞凋亡[15]。这些结果表明,Gli2可能是通过引起肾小管细胞凋亡而促进了AKI 中肾小管损伤。
近年来,Hedgehog/Gli 信号通路在肾纤维化中的作用倍受关注[16],为肾纤维化的防治提供了新的方向。研究表明,Hedgehog/Gli 信号通路的活化促进肾纤维化的形成。Hedgehog信号传导通路由Hh配体、膜蛋白受体、核转录因子和下游靶基因4部分组成。Hedgehog 通路起关键作用的核转录因子为Gli,其中Gli1和Gli2主要作为转录激活因子发挥功能,而Gli3主要作为转录抑制因子发挥功能[17-18],而且单独完全抑制Gli1 时仍不能控制纤维化,提示Gli2 是Hedgehog 通路致肾纤维化中重要的效应因子[16]。我们的研究也提示,沉默Gli2能下调纤维化、AKI标志物表达,并可下调Hedgehog通路蛋白受体Smo 表达。而过表达Gli2 可促进上调纤维化标志物。其他研究也表明Hedgehog/Gli2 通路在肾脏纤维化中的明确作用。可能的机制为Hedgehog 配体活化后对细胞膜上的受体发挥信号转导作用,并通过核转录因子Gli2影响肾小管上皮细胞周期阻滞,驱动肌成纤维细胞—细胞周期进展[11],导致肾小管上皮细胞损伤、修复不良及转变为肌成纤维细胞,作为纤维化的驱动因子,形成促纤维化微环境,加速肾纤维化形成和发展。而降低Gli2蛋白水平,可阻滞随后的肌成纤维细胞的细胞周期,从而减少肾脏纤维化。研究还认为,Gli2 抑制剂可能有望作为减少肌成纤维细胞增殖而缓解肾纤维化的治疗药物。这些均提示Hedgehog/Gli2 通路在肾损伤纤维化过程中发挥重要作用,Gli2 作为通路的转录因子可能是肌成纤维细胞增殖引起纤维化的关键干预靶点。
综上,本研究通过顺铂诱导AKI的体外细胞模型探讨Gli2 对AKI 中肾小管上皮细胞凋亡及纤维化的机制,研究结果提示Hedgehog/Gli2通路影响肾小管上皮细胞凋亡及纤维化转变,Gli2 可能在AKI发生发展中起重要作用,为阐明AKI后纤维化及慢性化转变的机制提供了一定理论依据,然本研究仅通过体外实验层面验证Gli2 在AKI 肾小管损伤及纤维化中的作用,尚无法阐明Hedgehog/Gli2通路在AKI 发生发展中的具体分子机制,有待未来开展更深入的研究探讨明确。
