蒺藜苜蓿(Medicago truncatula)叶片衰老的转录组分析
2021-11-09周玲芳尚骁尧张铁军晁跃辉
周玲芳, 尚骁尧, 张铁军, 晁跃辉
(北京林业大学草业与草原学院, 北京 100083)
衰老是一个细胞程序性死亡过程,受遗传因素和外界环境因子的调节。在植物叶片衰老过程中,叶肉细胞被解离和降解,营养物质被分解并运输到其他器官,并伴随生理生化指标、激素水平以及基因调控等方面发生一系列变化,以保证植物能完成正常的新陈代谢。叶绿素流失是衰老的特征之一,随着叶绿体分解,叶色褪绿变黄,并伴随总RNA[1]和蛋白质水平的下降。叶片衰老的过程非常复杂,涉及至少数千个基因的调控以及许多代谢和信号通路的改变。Lohman等[2]人将在衰老过程中表达上调的基因确定为衰老相关基因(Senescence associated genes,SAGs),这些基因通常编码实现细胞死亡的蛋白质[3],在植物衰老过程中起着重要的调控作用[4]。在分子遗传学研究中发现,植物叶片衰老相关基因主要包括光信号、激素转导和叶绿素代谢途径[5-6]。目前,人们已经在拟南芥(Arabidopsisthaliana)、大麦(HordeumvulgareL.)、番茄(Lycopersiconesculentum)、烟草(NicotianatabacumL.)等植物中发现了许多与衰老相关的基因[7]。Bayanati等[8]发现在玫瑰(Rosahybrida)衰老过程中,衰老相关因子可以调控衰老相关基因RhAA和RhCG的表达,与衰老的降解和再动员过程有关。在葡萄(VitisviniferaL.)[9]和拟南芥[10]的衰老过程中,SAG12转录水平显著增加,表达上调。SAG113定位于高尔基体,编码一种属于PP2C家族的蛋白磷酸酶。在衰老的叶片中,ABA会诱导SAG113基因表达,从而抑制气孔关闭,导致水分流失,加速衰老进程[11]。转录因子(Transcription factors,TFs)通过与靶标基因启动子区域的顺式作用元件结合并作用,调控靶基因的表达,在植物生长发育、激素信号传导和新陈代谢等多方面中发挥重要作用[12]。目前,人们在高等植物中已经发现了许多类型的转录因子,共同作用调节叶片衰老过程中的基因表达。如NAP是近来年发现的植物中特有的一类转录因子,属于NAC家族。李钰卓等[13]从菜薹(Brassicarapavar.parachinensis)中分离BrNAP1基因并分析其功能,发现BrNAP1是菜薹采后叶片衰老的正调控因子,受ABA诱导表达上调,且能够激活BrSAG113表达。在矮牵牛花(Petuniahybrida)的花冠和雌蕊中对ERF基因进行了表达分析,这些基因在花衰老过程中表现出不同的表达模式和水平,并且都能被外源乙烯加速转录,表明PhERF参与了牵牛花衰老的转录调控过程[14]。WRKY转录因子能够特异性识别W-box结合元件并产生相互作用,进而参与植物生长发育调控衰老[15]。在拟南芥等模式植物中已经证实WRKY转录因子具调控叶片衰老作用,GhWRKY33转基因的拟南芥与野生型相比,衰老延缓,并且衰老相关基因AtSAG12、AtSAG13的表达下调,表明GhWRKY33是调控拟南芥衰老的负调控因子[16]。
转录组测序称RNA-Seq,利用高通量测序技术进行分析,可全面快速地获得植物特定细胞或组织在某个状态下几乎所有的转录本及基因序列,在新基因的发现和基因表达分析方面发挥了重要作用[17]。近年来,Illumina测序广泛应用于研究植物的生长发育、抗逆境生理机制以及挖掘基因功能等[18-20]。目前,有许多植物物种通过测序技术开展了叶片衰老的分析,如烟草[21]、葡萄[22]和高羊茅(Festucaarundinacea)[23]等。然而,在蒺藜苜蓿中有关叶片衰老的研究还较少,转录调控机制尚不明确。
蒺藜苜蓿(Medicagotruncatula)是一种重要的豆科牧草植物,由于其生长周期短、倍性小(2n=16)、基因组小和自花授粉等优点成为了研究豆科植物的模式植物[24]。R108品种由于其较高的遗传转化效率,被广泛应用于蒺藜苜蓿的分子生物学与遗传学研究[25]。目前蒺藜苜蓿已完成全基因组测序,为有效地研究基因调控、深度挖掘其潜藏的遗传信息提供了一定依据。叶片衰老是植物健康生长、繁育后代的关键指标之一,对作物的产量和品质有很大影响。因此,深入研究叶片衰老的机理,人为调控叶片衰老过程对培育优良品系及提高作物产量具有重要的理论意义和实践价值。
本研究以蒺藜苜蓿健康、成熟及3个不同衰老阶段叶片为材料,利用RNA-Seq手段进行转录组分析,探究叶片衰老的分子调控机制,筛选差异表达基因(Differentially expressed genes,DEGs)并进行功能注释,初步鉴定SAGs和TFs,为进一步研究叶片衰老相关基因的功能提供基础。
1 材料与方法
1.1 植物材料及RNA提取
试验材料为蒺藜苜蓿R108品种,由北京林业大学草坪科学与管理实验室保存。将蒺藜苜蓿种子用75%乙醇灭菌10分钟后,置于湿润滤纸上,等2~3周长出幼苗后转移至含草炭土、蛭石、珍珠岩(1:1:1)的营养土中,放置于气候培养箱中,25℃光照16 h/22℃黑暗8 h。选取长势健康良好的蒺藜苜蓿植株,采集其成熟叶片以及衰老初、中、末期叶片,在液氮中速冻后置于-80℃冰箱中保存以备后续实验。
利用Plant RNA Kit试剂盒(OMEGA公司)提取上述样本叶片的总RNA,用1%琼脂糖凝胶电泳检测RNA的降解程度和污染情况,并结合超微量紫外分光光度计(OD260/280)检测其纯度和浓度,然后置于-80℃冰箱中保存备用。
1.2 叶绿素含量的测定
叶片衰老显著的形态特征是叶片颜色的变化,而叶片变黄是鉴定叶片衰老开始和发展的可见依据,根据蒺藜苜蓿叶片的黄化程度,将衰老过程分为3个阶段,分别是衰老初期、衰老中期和衰老后期[26]。在本研究中,叶片完全展开并且没有变黄的迹象被定义为成熟阶段,并对每个阶段的叶片进行叶绿素含量的测定。取新鲜成熟和衰老初、中、末期叶片样品各0.5 g,分别加入95%乙醇25 mL,在黑暗条件下室温提取24~36 h后,使用紫外可见分光光度计测量665 nm,649 nm处的吸光度,并通过公式计算叶绿素含量。
1.3 文库构建及测序质量分析
通过Qubit对RNA浓度进行精确定量,Agilent 2100 Bioanalyzer精确检测RNA完整性后,经北京诺禾致源科技公司进行转录组测序分析。样品检测合格后,用带有Oligo(dT)的磁珠,通过A-T互补配对与mRNA的ployA尾结合的方式富集真核生物的mRNA,最后进行PCR富集得到最终的cDNA文库。
文库构建完成后,先使用Qubit2.0进行初步定量,稀释文库至1 ng·μL-1,随后使用Agilent 2100对文库的插入片段长度(Insert size)进行检测,Insert size符合预期后,使用q-PCR方法对文库的有效浓度进行准确定量(文库有效浓度>2 nM),以保证文库质量。库检合格后在Illumina HiSeq平台上进行测序。
为了保证信息分析质量,对得到的原始序列数据(Raw reads)进行过滤,去除里面含有带接头的(Adapter)、N比例大于10%以及低质量的reads,得到过滤后序列数据(Clean reads)。利用Bowtie (2.2.3)[27]建立参照基因组索引,使用TopHat(2.0.12)[28]将Clean reads与参考基因组进行比对。采用HTSeq(0.61)软件计算每个基因的读取数[29],然后根据基因的长度计算每个基因的FPKM(Expected number of Fragments Per Kilobase of transcript sequence per Millions base pairs sequenced,每百万fragments中来自比对到某一基因每千碱基长度的fragments数目),并将读取计数映射到该基因上。
1.4 差异表达基因功能注释
利用DEseq软件(1.18.0)[30]对成熟和衰老叶片样本进行基因差异表达分析,然后根据模型进行假设检验概率(Pvalue)的计算进行多重假设检验校正,控制错误发现率(False discovery rate,FDR)。差异表达基因以|log2(Fold Change)|>1且FDR<0.05作为筛选标准。
通过NCBI Blastx比对获得差异表达基因的Nr信息,得到最高序列相似性的蛋白,从而得到基因的Nr注释;利用GOseq R包(Release 2.12)[31]对差异表达基因进行GO富集分析;利用KOBAS软件(2.0)[31]检测KEGG通路中DEGs的数据富集情况。
1.5 转录因子分析和衰老相关基因筛选
从PlnTFDB(http://plntfdb.bio.uni-potsdam.de/v3.0/)下载植物转录因子数据库,利用Blastx将所得到的差异表达基因序列与植物转录因子数据库的序列进行比对。
通过使用Blastx将差异表达基因和叶片衰老数据库(Leaf senescence database,LSD)中的基因序列进行比较,分析获得蒺藜苜蓿与其他植物之间具有较高同源性的衰老相关基因。
1.6 qRT-PCR验证
为了验证差异表达基因的结果,随机选择8个差异表达基因,通过qRT-PCR进行测量。利用Primer Premier 5.0软件设计引物(表1)。根据TB GreenPremixExTaqTMⅡ试剂盒(Takara公司)说明书进行操作。20 μL反应体系为TB GreenPremixExTaqII 10 μL,上下游引物各1 μL,cDNA 1 μL,RNase Free H2O 7 μL。使用2-△△Ct法[33]计算基因相对表达量,所有基因的表达分析均设置了3次生物学重复。
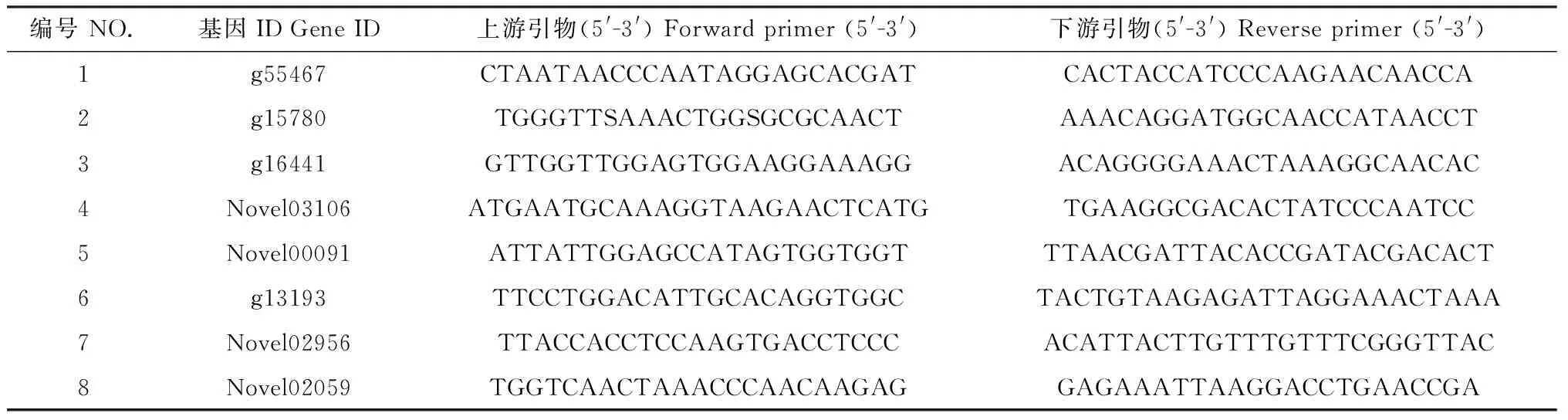
表1 qRT-PCR引物信息Table 1 List of primers used for qRT-PCR
2 结果分析
2.1 测序数据分析
如图1所示,衰老初期、中期和末期叶片的叶绿素含量分别下降到成熟叶片的20.72%,15.49%和8.0%。表明样品可以反映叶片衰老的进程,可用于衰老相关基因的鉴定。
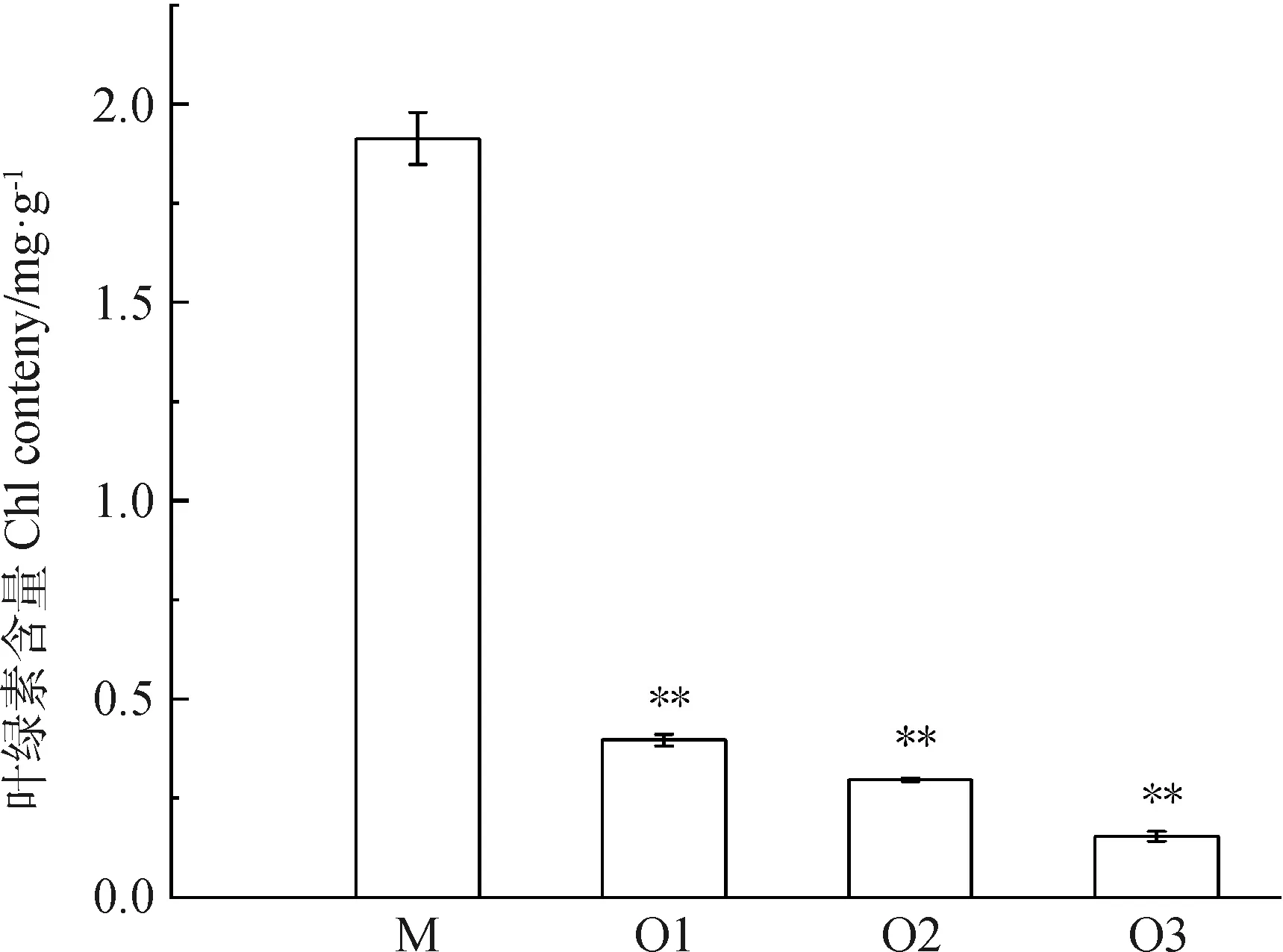
图1 叶绿素含量Fig.1 Chlorophyll content in leaf samples注:M:成熟叶片;O1:衰老初期叶片;O2:衰老中期叶片;O3:衰老末期叶片;**表示0.01水平下有显著性差异Notes:M:Mature leaf;O1:Early senescent leaf;O2:Medium senescent leaf;O3:Late senescent leaf;** indicate significantly different at the 0.01 probability levels,respectively
本研究以蒺藜苜蓿成熟叶片和衰老初、中、末期叶片为样品进行建库和转录组测序,测序数据质量情况如表2所示。原始序列(BioProject号:PRJNA754763)过滤掉低质量的reads后,共计获得82.81 Gb的Clean reads,且每个文库的Clean reads均在41万条以上,其中Q20均大于 97%和Q30(FDR≤0.1%)均大于93%,GC含量为41.44%~42.68%,表明转录组测序质量较好,可以进行下一步分析。

表2 转录组测序数据质量分析Table 2 Quality analysis of transcriptome sequencing data
2.2 差异表达基因鉴定
在4个发育阶段的叶片中共鉴定出64 324个基因。通过DEGs分析,以成熟叶片样品为对照,衰老初期的样品中,有6 746个基因上调表达,6 555个基因下调表达;衰老中期的样品中,有7 853个基因上调表达,7 546个基因下调表达;衰老末期样品中,有7 544个基因上调表达,7 304个基因下调表达(图2)。为了进一步分析在衰老过程中的调控基因,筛选3个衰老阶段均差异表达的基因,共鉴定出2 990个差异表达基因,其中上调基因1 362个,下调基因1 628个。对这2 990个差异表达基因进行长度分析,结果显示其中2 393个基因长度大于500 bp,占80.03%(图3)。

图2 差异表达基因火山图Fig.2 Volcano plot of differentially expressed genes注:A:衰老初期叶片vs成熟叶片;B:衰老中期叶片vs成熟叶片;C:衰老末期叶片vs成熟叶片Notes:A:Early senescent leaf vs Mature leaf;B:Medium senescent leaf vs Mature leaf;C:Late senescent leaf vs Mature leaf
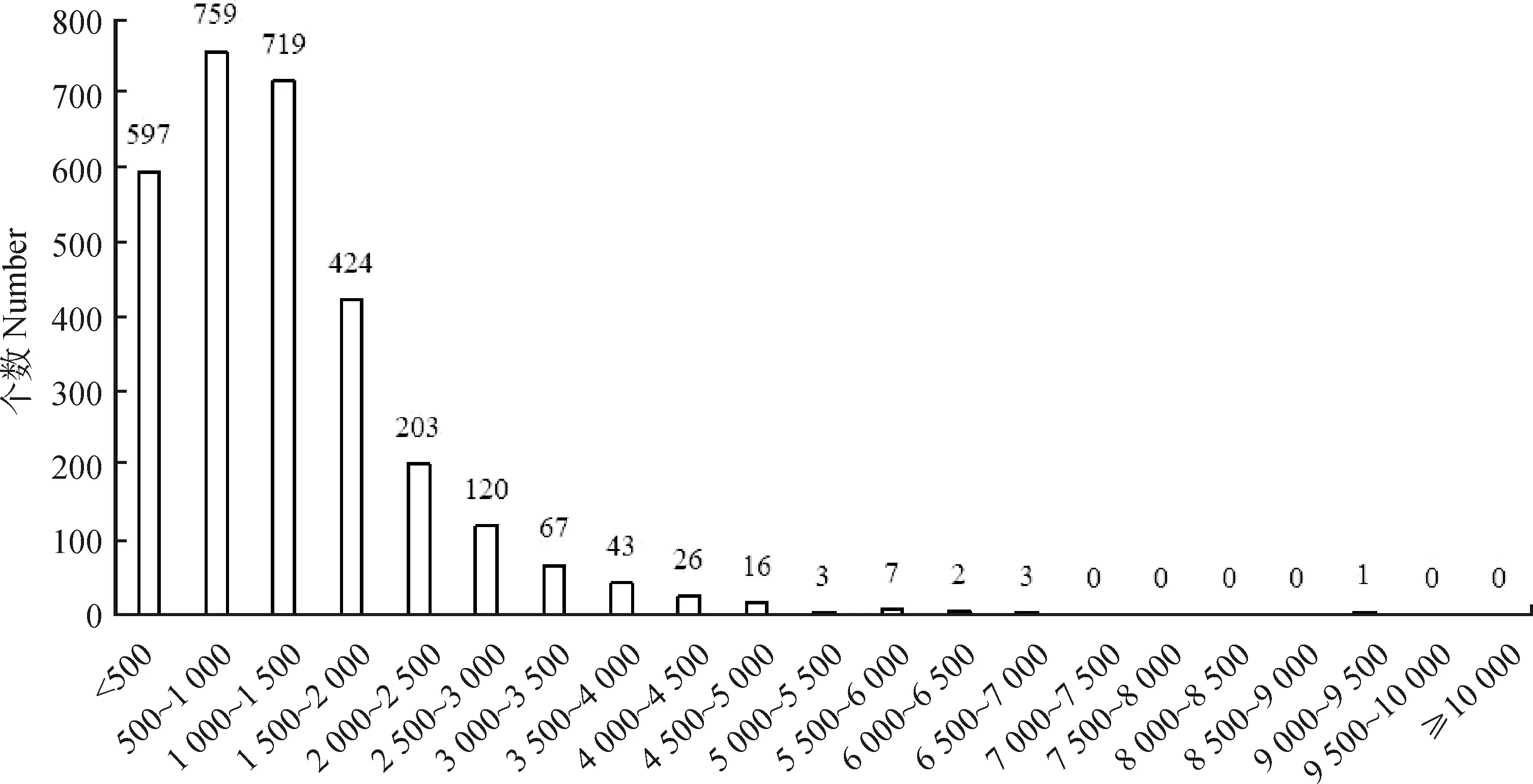
图3 差异表达基因长度分布Fig.3 Length distribution of differentially expressed genes
2.3 差异表达基因功能注释
为了探究差异表达基因的功能信息,通过BLAST软件将筛选出来的2 990条差异表达基因与COG,GO,KEGG,KOG,Pfam,Swissport以及Nr数据库进行比对,能够得到注释信息的差异表达基因一共有2 684条。其中,注释到Nr数据库的数量最多,有2 376条占所有差异表达基因的79.46%。注释到COG数据库的数量最少,仅有724条,占所有差异表达基因的24.21%。(表3)

表3 差异表达基因注释统计Table 3 DEGs annotation statistics
2.4 差异表达基因功能富集分析
将所有差异基因同Nr数据库进行比对,结果显示数量分布最多的前5物种分别为:蒺藜苜蓿Medicagotruncatula(2 233个),鹰嘴豆Cicerarietinum(37个),葡萄Vitisvinifera(12个),野大豆Glycinesoja(11个),大豆Glycinemax(10个)。
通过GO数据库对差异表达基因进行功能注释以及富集分析显示,有938条差异表达基因被注释,基因功能主要分为3个方面:细胞组分、分子功能以及生物过程(图5)。其中,注释到生物过程方面的差异表达基因最多,共有1 530个分布在17个亚类中,主要富集于代谢过程、细胞过程和单一生物过程;其次是注释到分子功能方面共11个亚类的基因有1 461个,主要集中于催化活性、结合和核酸结合转录因子活性;注释到细胞组分方面共12个亚类的基因有457个,主要富集于细胞、细胞组分、膜和细胞器等。
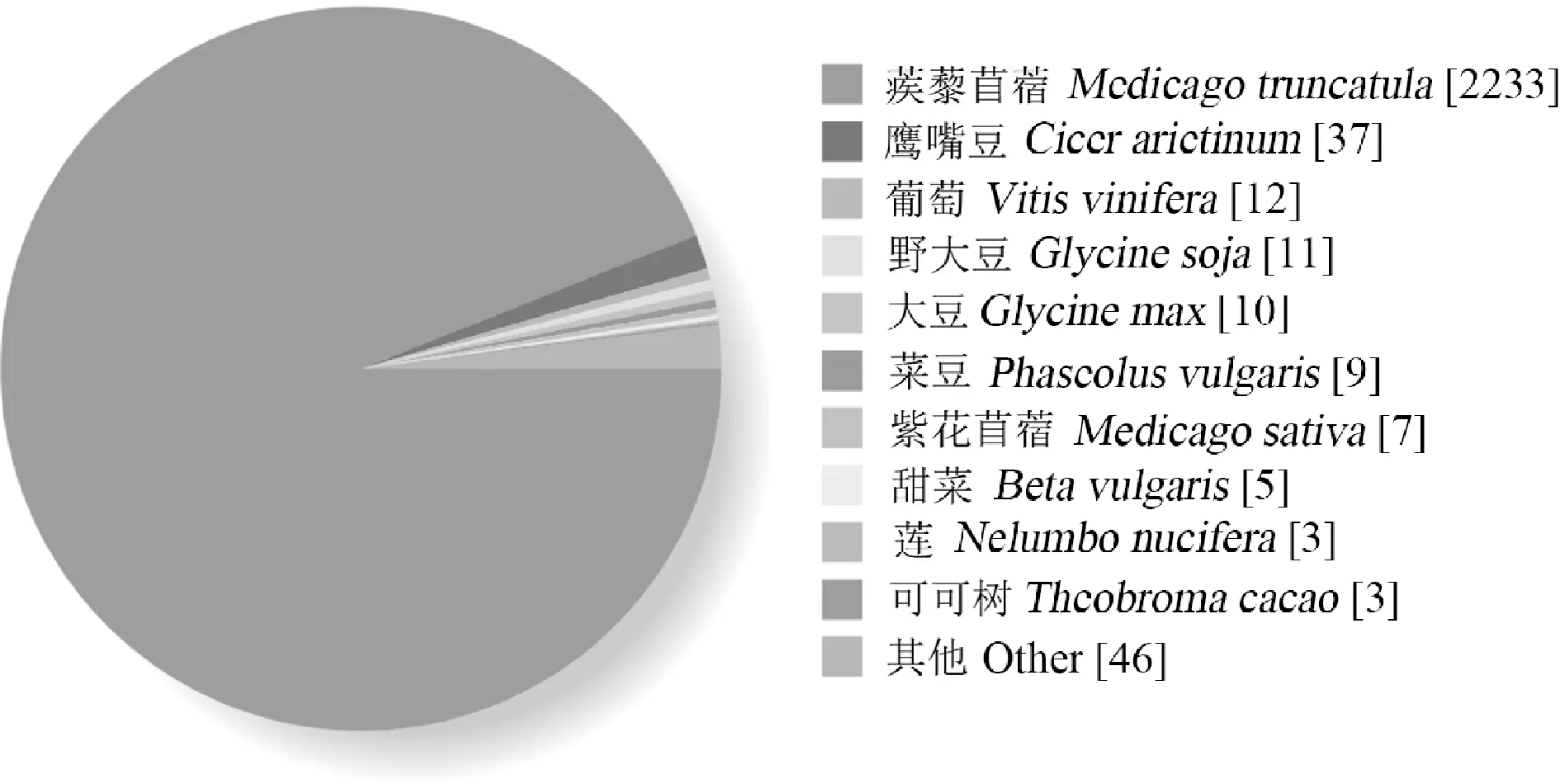
图4 Nr同源物种分布Fig.4 Nr Homologous Species Distribution

图5 差异表达基因GO功能分类Fig.5 Gene ontology function classification of Differentially expressed genes
通过COG数据库对差异表达基因进行比对,发现有724条基因得到注释,分配至26类功能中,且基因在不同的功能分类中存在明显的差异。其中,糖运输和新陈代谢功能类别注释的基因最多,其次是一般功能预测类和信号转导机制,之后是翻译后修饰、蛋白折叠和分子伴侣。

图6 COG功能分类统计Fig.6 COG function classification statistics
将筛选出来的差异基因与KEGG数据库进行比对以分析代谢途径,结果显示共有353个差异基因成功富集于83个代谢途径通路,注释率为11.8%(表4)。集中在植物激素信号转导(Plant hormone signal transduction)和苯丙烷类生物合成(Phenylpropanoid biosynthesis)的差异表达基因数量较多,分别有24个和16个,其次是淀粉和蔗糖代谢(Starch and sucrose metabolism)有13个。表明蒺藜苜蓿叶片在衰老过程中,多种植物激素含量、生物合成途径以及代谢途径均发生变化,参与这些途径的差异基因发挥了重要作用。
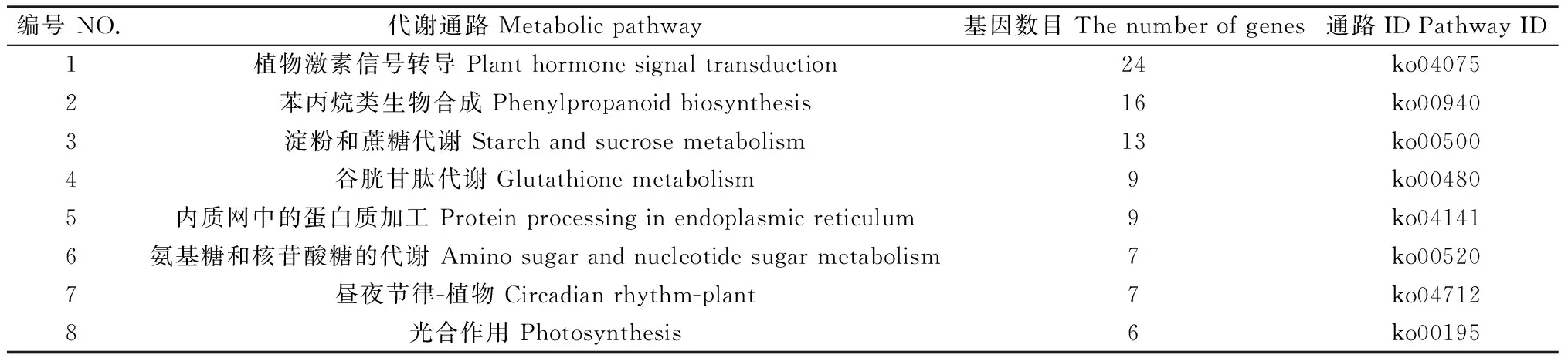
表4 差异表达基因的部分KEGG通路Table 4 Partial KEGG analysis of Differentially expressed genes
2.5 基因转录因子分析
通过与植物转录因子数据库比对,共鉴定出145个转录本,分布在33个转录因子家族中,占主体的6个家族是ERF(20个)、WRKY(18个)、bHLH(16个)、MYB(12个)、NAC(9个)和bZIP(7个),其中63个转录因子在衰老叶片中显著上调(图7)。
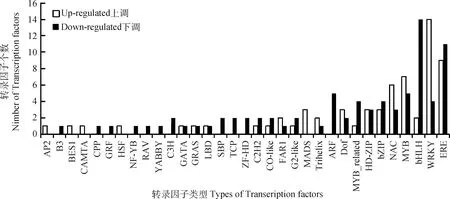
图7 转录因子家族分布Fig.7 Transcription family distribution
2.6 衰老相关基因分析
LSD是一个综合性资源包括衰老相关基因及其突变体。将筛选出来的差异表达基因与LSD中的序列进行比对,分析蒺藜苜蓿与其他植物之间衰老相关基因序列的保守性。2 990个差异表达基因中,有837个在LSD中具有其他植物的同源基因,其中393个基因表达上调,444个基因表达下调,主要涉及转录调控、激素反应、信号转导、脂质或碳水化合物代谢、蛋白质降解或修改,营养循环和其他(表5)。
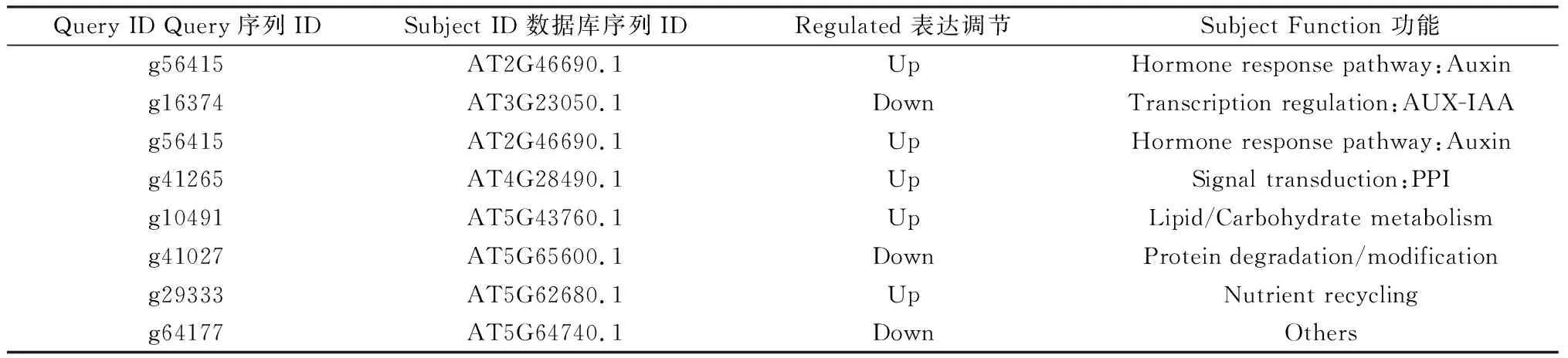
表5 蒺藜苜蓿与其它物种叶片衰老的部分同源物Table 5 Partial list of leaf senescence orthologs between Medicago truncatula and other species
2.7 qRT-PCR验证
为验证蒺藜苜蓿衰老叶片转录组测序结果的准确性,随机选择8个差异基因并设计引物,其中4个上调基因为g55467,g15780,g16441和Novel03106,4个下调基因为Novel00091,g13193,Novel02956和Novel02059,进行实时荧光定量表达分析。结果表明这8个差异基因在qRT-PCR与转录组测序中显示出相似的表达模式(图8)
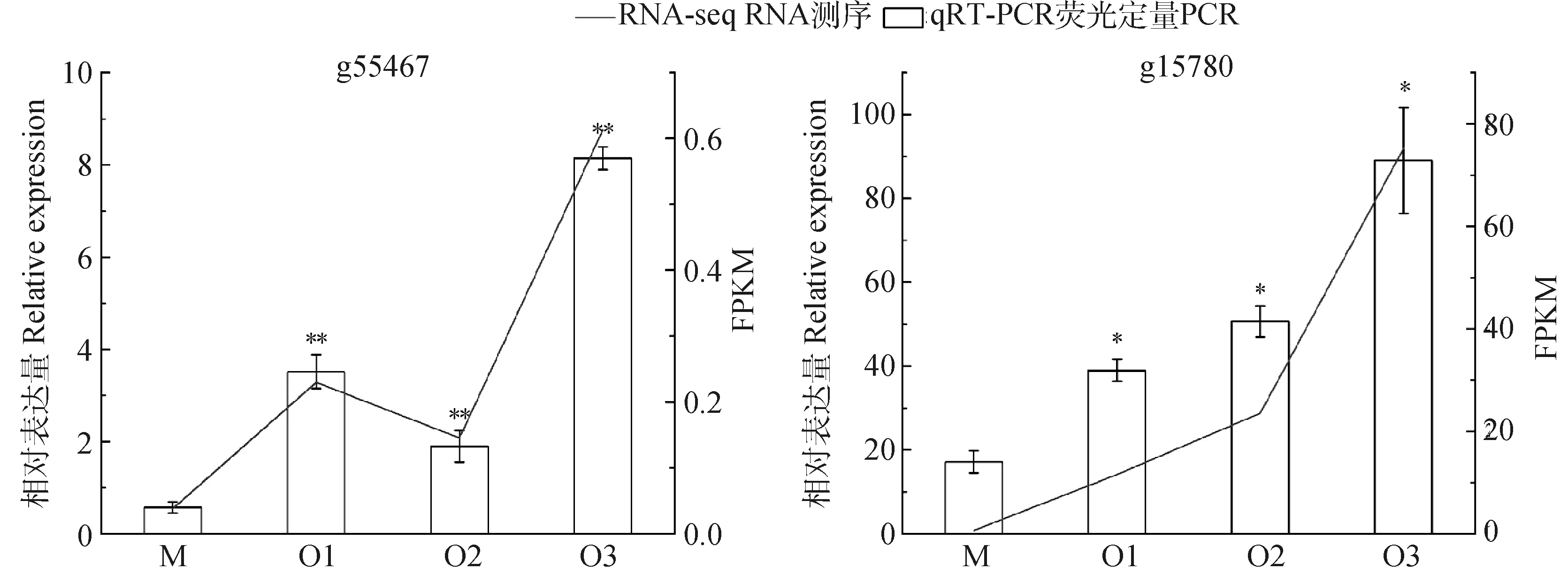
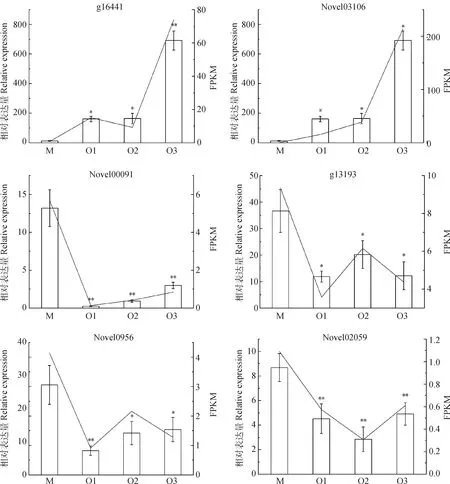
图8 差异表达基因qRT-PCR验证Fig.8 qRT-PCR verification for differentially expressed genes注:M:成熟叶片;O1:衰老初期叶片;O2:衰老中期叶片;O3:衰老末期叶片;*与**分别表示在 0.05和0.01水平下有显著性差异Notes:M:Mature leaf;O1:Early senescent leaf;O2:Medium senescent leaf;O3:Late senescent leaf;* and ** indicate significantly different at the 0.05 and 0.01 probability levels,respectively
3 讨论
近年来,高通量测序技术在植物的研究上应用日益广泛,受到众多研究者的青睐。2014年R108蒺藜苜蓿被测序成功,为研究蒺藜苜蓿在不同发育时期基因表达提供了重要参考。本研究为获得更多的转录组测序信息,将叶片衰老分为3个阶段,以蒺藜苜蓿成熟叶片和衰老初、中、末期叶片为材料,采用Illumina HiSeq技术进行转录组测序并进行相关分析,共计获得82.81 Gb的Clean reads且Q20和Q30均大于90%,说明测序数据质量较好,可用于进一步研究。
通过转录组鉴定发现在叶片衰老的过程中共有2990个基因差异表达,其中表达下调基因较多,结果与以前的报告一致,用衰老叶片组织构建的cDNA文库进行筛查,在上、下调2种特异表达的基因中,多数基因为下调表达[34]。目前利用转录组分析,也筛选出很多其他植物在衰老过程中的差异表达基因。如Lu等对毛果杨(Populustrichocarpa)在季节性衰老过程中的转录组变化进行分析,共鉴定出17 974个差异表达基因[35];Chao等利用高通量Illumina测序进行分析,得出红三叶(TrifoliumpratenseL.)衰老和非衰老叶片之间有481个基因差异表达[36]。
基于Blast分析,2 684条差异表达基因通过Nr、KEGG、GO等七个数据库比对被成功注释,仍有306个基因未被注释,推测是存在新基因,或者是由于其他的污染,如实验采样过程中样品被未知微生物污染。在GO分类结果表明,这些差异表达基因在叶片衰老过程中,主要集中在生物过程的代谢过程、细胞过程和单一生物过程。此外,基于KEGG富集分析,我们发现差异基因在植物激素信号转导和苯丙烷类生物合成显著富集,以前的研究也表明在油茶(CamelliaoleiferaAbel.)叶片衰老过程中,植物激素信号转导和苯丙烷类生物合成表现出最多的差异表达基因[37]。
叶片衰老的过程受到基因的调控,其中转录因子发挥了重要作用,因此我们对转录因子进行了统计分析,发现差异表达的转录因子主要分布在ERF、WRKY、bHLH、MYB、NAC等家族中。目前很多研究鉴定出拟南芥中有大量的WRKY转录因子与叶片衰老密切相关,除AtWRKY6 在拟南芥叶片衰老期间的表达量快速上调外[38],拟南芥WRKY53作为叶片衰老的核心转录因子,诱导其表达也可以加速叶片衰老[39]。本研究中,发现18个WRKY转录因子基因与叶片衰老有关,其中有14个基因在衰老过程中表达上调,这与前人的研究结果一致。此外,我们发现有6个NAC转录因子基因上调表达,参与调控蒺藜苜蓿叶片衰老过程。有研究表明,与调控衰老有关的NAC转录因子大多是以正调控的方式在叶片衰老过程中发挥重要作用[40-41]。 Zhang等[42]通过研究发现,转录因子AtNAP在衰老的拟南芥中表达,并且在衰老叶片中NAP转录因子通过ABA-AtNAP-SAG113 PP2C调节链,控制叶片气孔运动从而促进衰老;而OsNAP作为AtNAP在水稻(OryzasativaL.)中的同源基因,降低OsNAP基因表达可显著延缓水稻叶片衰老[43],以上证实了一些NAC转录因子可以直接或间接的加速叶片衰老。
4 结论
本研究基于蒺藜苜蓿成熟叶片与衰老初期、中期、末期叶片的转录组测序获得的大量数据,共筛选出2 990个差异表达基因,其中1 362个基因显著上调,1 628个基因显著下调。通过后续对差异表达基因进行功能注释以及分析转录因子等初步探索了蒺藜苜蓿叶片衰老的分子调控机制,为今后进一步挖掘豆科植物叶片发育和衰老的调控机制提供参考。
