大叶落地生根酵母cDNA文库的构建及KdSAHH基因自激活检测
2021-11-09王梦迪胡乃文晁跃辉曾会明
张 珂, 王梦迪, 董 笛, 胡乃文, 晁跃辉, 曾会明
(北京林业大学草业与草原学院,北京 100083)
大叶落地生根是景天科伽蓝菜属多年生肉质草本植物,不定芽入土即可形成新的植株,故名“落地生根”,原产于非洲马达加斯加,广泛分布于热带和亚热带地区[1]。大叶落地生根具有较高的研究价值,它不仅是研究景天酸代谢(Crassulacean acid metabolism,CAM)途径的模式植物[2],也是研究高等植物无性繁殖的模式植物[3]。大叶落地生根叶缘凹陷处会形成可进行无性繁殖的不定芽,这一特质可用来研究植物体细胞全能性,为植物组织培养等相关应用的发展提供理论基础[4]。
随着功能基因组学研究的逐步深入,蛋白互作成为分子生物学领域的研究热点,研究蛋白质之间的相互作用是研究基因功能的重要方法[5]。酵母杂交技术是高灵敏度的分子生物学技术,可以在酵母体内分析蛋白质互作或蛋白质与核酸互作[6]。同时,该技术既能免去纯化蛋白质复杂操作且在一定程度上还能反应细胞内的真实情况[7]。酵母杂交技术已广泛应用于蛋白质互作方面的研究,用以揭示已知蛋白或基因参与调控动植物特定代谢过程的分子机制。赵倩倩等[8]构建了玉米不同时期籽粒cDNA文库,并从中初步筛选到了玉米籽粒灌浆期重要调控蛋白ZmSCL1的互作因子。姜红岩等[9]探究ZjNAC2在胁迫响应中的作用机制时,成功从日本结缕草酵母杂交文库中筛选到4个与抗病过程相关的互作蛋白。酵母杂交技术的关键在于构建高质量的基因文库。因此,构建高容量的cDNA酵母文库对酵母杂交的筛选具有重要意义。目前,对于大叶落地生根不定芽的研究集中在功能基因的克隆及表达分析方面[10-11],尚未见利用酵母杂交技术探究其分子调控机理的研究。
S-腺苷高半胱氨酸水解酶(S-adenosyl-homocysteine hydrolase,SAHH)是调节细胞内甲基化反应的关键酶。SAHH基因影响着植物的生长发育,抑制该基因的表达会导致植物器官的发育缺陷,如烟草花和叶的畸变[12]。本课题组[13]前期在干旱诱导的大叶落地生根不定芽差减cDNA文库中筛选到KdSAHH基因片段,并利用荧光定量技术发现KdSAHH基因在不定芽发育过程中上调表达明显。为探究KdSAHH基因在大叶落地生根不定芽发育过程中分子调控机制,本研究利用SMART技术构建了高容量大叶落地生根酵母杂交cDNA文库和pGBKT7-KdSAHH诱饵载体,以期为利用酵母杂交技术从大叶落地生根cDNA文库中筛选出KdSAHH的互作因子,揭示KdSAHH基因调控下游响应基因从而影响不定芽发生的分子机制提供研究基础。
1 材料与方法
1.1 试验材料
大叶落地生根培养于北京林业大学草坪研究所光照培养箱,选择株龄为1年且不定芽生长稳定的大叶落地生根为试验材料。RNA提取试剂盒、PCR纯化试剂盒、质粒提取试剂盒均购自OMEGA公司。反转录试剂盒、SmaⅠ限制性内切酶、无缝连接酶均购自TaKaRa公司(日本)。文库构建试剂盒、酵母转化试剂盒、Y187菌株、酵母表达载体pGADT7,Y2HGold酵母菌株、酵母诱饵载体pGBKT7,X-α-Gal,AbA,Matchmaker Insert Check PCR Mix 1均购自Clontech公司。大肠杆菌感受态E.coli DH5α购自Transgen公司。
1.2 大叶落地生根总RNA的提取
选取大叶落地生根长势良好且均一的根、茎、叶、花以及不定芽组织混匀,称取0.1 g于液氮中研磨至粉末状,采用试剂盒提取总RNA。用1%琼脂糖凝胶电泳检测总RNA的完整性,并用紫外分光光度计NanoDorp 2000测量其质量与浓度。
1.3 cDNA的合成
1.3.1cDNA第一链合成 取1 μg RNA为模板,与1 μL CDS Ⅲ Primer Mix混匀并加水至4 μL体系中72℃温浴2 min,冰浴2 min后加入2 μL 5×第一链缓冲液,1 μL DTT,1 μL dNTP Mix,1 μL SmartscribeTM反转录酶,然后置于42℃保温10 min,加入1 μL SMART III寡核苷酸,42℃保温1 h,然后加入1 μL RNase H,37℃保温20 min。将一链产物-20℃保存。
1.3.2LD PCR 取2 μL cDNA第一链产物和10 μL 10×Advantage 2 PCR缓冲液,2 μL 50×dNTP Mix,2 μL 5′ PCR引物,2 μL 3′ PCR引物,2 μL 50×Advantage 2 Polymerase Mix加水至100 μL体系中混匀。5′PCR引物:5′-TTCCACCCAAGCAGTGGTATCAACGCAGAGTGG-3′,3′ PCR引物:5′-GTA-TCGATGCCCACCCTCTAGAGGCCGAGGCGGCCGACA-3′。PCR反应程序为:95℃ 30 s;95℃ 15 s,68℃ 6 min,18个循环;68℃ 5 min。然后利用PCR纯化试剂盒纯化其产物。
1.4 cDNA文库的均一化
1.4.1杂交 取1 μg ds cDNA,8 μL 2×杂交反应缓冲液加水至16 μL体系的PCR小管中混匀后均分4份,置于PCR仪中98℃保温2 min后,迅速转移到68℃水浴锅中5 h。
1.4.2DSN消化 68℃预热2×DSN主缓冲液。取4 μL杂交后的cDNA,5 μL预热2×DSN主缓冲液和1 μL DSN溶液(分别为DSN,1/2 DSN,1/4 DSN,DSN储存缓冲液)于4个PCR小管混匀,置于PCR仪中68℃保温25 min,每管加入10 μL 2×DSN终止缓冲液后室温放置5 min,将产物-20℃保存。
1.5 DSN消化后两次PCR放大
1.5.1第一次放大 取5 μL 10×Advantage 2 PCR缓冲液,1 μL 50×dNTP Mix,5 μL 10×GC溶解溶液,1 μL 5′ PCR引物,1 μL 3′ PCR引物,1 μL 50×Advantage 2 Polymerase Mix和1 μL消化后产物加水至50 μL体系的PCR小管中混匀,并分别标记为DSNP1,1/2 DSNP1,1/4 DSNP1,DP1。PCR反应程序为:95℃ 30 s;95℃ 15 s,68℃ 6 min,17个循环;68℃ 5 min。用1%琼脂糖凝胶电泳检测后选择模板进行第二次放大。
1.5.2第二次放大 取10 μL 10×Advantage 2 PCR缓冲液,2 μL 50×dNTP Mix,10 μL 10×GC溶解溶液,2 μL 5′ PCR引物,2 μL 3′ PCR引物,2 μL 50×Advantage 2 Polymerase Mix和2 μL稀释十倍后一次放大产物加水至100 μL体系的PCR小管中混匀。PCR反应程序为:95℃ 30 s;95℃ 15 s,68℃ 6 min,24个循环;68℃ 5 min。用1%琼脂糖凝胶电泳检测,然后利用纯化试剂盒纯化PCR产物。
1.6 大叶落地生根cDNA酵母文库构建与克隆鉴定
按照酵母转化试剂盒说明书制备Y187酵母感受态,并参照赵倩倩等[8]酵母共转化的方法转化Y187酵母感受态细胞。吸取200 μL转化后的酵母细胞液铺展到规格为直径150 mm的平板上,共计100块平板。30℃倒置温育平板直到克隆出现。4℃冷却平板3~4 h,每个平板加入5 mL冷冻培养基,使用无菌的玻璃棒并轻轻的搅动,收集液体。吸取100 μL 10倍、100倍和1 000倍的酵母稀释液铺展到在规格为直径100 mm SD/-Leu的平板上检测转化效率。文库滴度(CFU·mL-1)=平板上的单克隆个数/平板上涂布菌液的体积(μL)×1×103(μL),文库总容量=文库滴度(CFU·mL-1)×文库菌液总体积(mL)。随机挑取24个单克隆菌落,使用Matchmaker Insert Check PCR Mix 1进行PCR检测。上游引物序列T7:5′-TAATACGACTCACTATAGGG-3′,下游引物序列3′AD:5′-AGATGGTGCACGATGCACAG-3′。PCR反应程序为:95℃ 1 min;98℃ 10 s,55℃ 30 s,68℃ 2 min,30个循环。用1%琼脂糖凝胶电泳检测插入片段大小。
1.7 pGBKT7- KdSAHH载体的构建及Y2H Gold酵母转化
根据KdSAHH基因序列(GenBank登录号:KF953475)设计引物。上游引物序列pGBKT7-KdSAHH-F:5′-ATGGCCATGGAGGCCGAATTCG-CGCTTATCGTCGAGAAAAC-3′,下游引物序列pGBKT7-KdSAHH-R:5′-CTGCAGGTCGACGG-ATCCCCTCAGTACCTGTAGGCAGCAG-3′。提取实验室保存测序结果正确的大肠杆菌菌液的质粒进行PCR扩增。PCR反应程序为:98℃ 10 s;98℃ 10 s,68℃ 70 s,30个循环;72℃ 5 min。pGBKT7载体用SmaⅠ单酶切后与PCR产物一同纯化回收并用无缝连接酶进行载体与PCR产物的连接。将连接产物转化至DH5α大肠杆菌感受态中,37℃倒置培养。挑取单菌落进行PCR及琼脂糖凝胶电泳检测,将检测正确的菌液送至生物公司测序。提取测序正确的pGBKT7-KdSAHH重组质粒,并将其转化至Y2HGold酵母感受态中。
1.8 诱饵蛋白自激活检测
挑取转化成功的酵母单菌落培养于SD/-Trp液体培养基中,30℃过夜培养,取10 μL 10倍、100倍和1 000倍酵母稀释液滴至SD/-Trp,SD/-Trp/X-α-Gal和SD/-Trp/X-α-Gal/AbA固体培养基上,30℃倒置培养,观察并记录酵母生长情况和生长状态。
2 结果与分析
2.1 大叶落地生根总RNA的提取
将提取的总RNA用1%琼脂糖凝胶电泳检测。结果显示(图1),28S和18S条带清晰明亮,完整性好。紫外分光光度计NanoDorp 2000测量浓度为300 ng·μL-1,A260/A280值为2.11,A260/A230值为2.01,表明RNA质量好、纯度高,无蛋白、酚类物质和有机溶剂等的污染,满足建库要求。
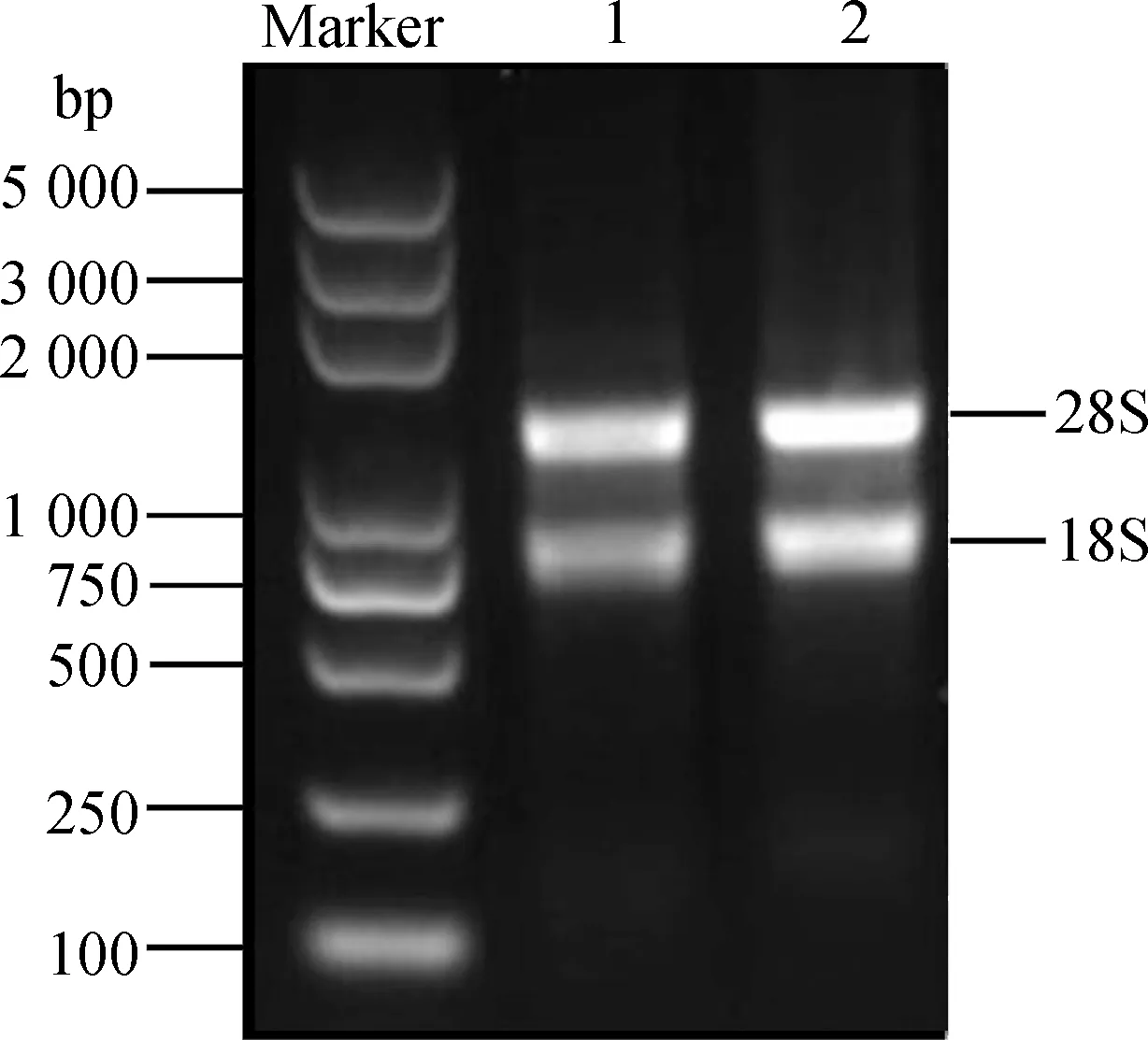
图1 总RNA琼脂糖凝胶电泳Fig.1 Agarose gel electrophoresis of total RNA注:Marker为Trans2K Plus DNA Marker;1和2为总RNANote:Marker indicate Trans2K Plus DNA Marker;1 and 2 indicate total RNA
2.2 双链cDNA文库的构建及均一化处理
以1 μg RNA为模板利用SMART技术反转录成单链cDNA,再以其为模板进行LD-PCR合成双链cDNA。对双链cDNA进行用1%琼脂糖凝胶电泳检测,结果显示(图2A),双链cDNA条带主要分布在750~3 000 bp之间,且条带成弥散状。将质量良好的双链cDNA进行文库均一化处理并经DSN消化后两次PCR放大处理。结果显示(图2B),处理后的双链cDNA条带均一且呈弥散状,表明均一化效果好,可用于后续试验。
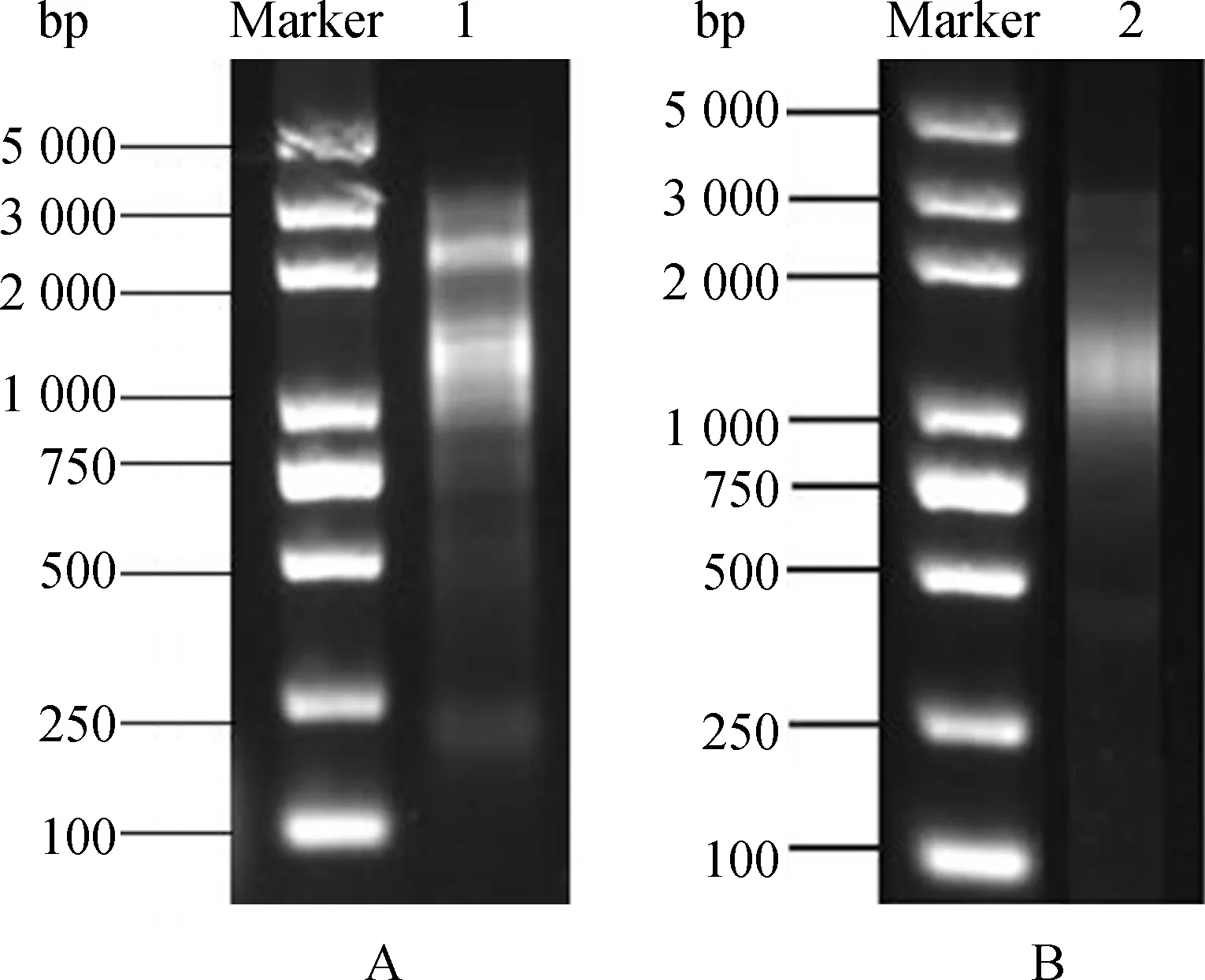
图2 双链cDNA琼脂糖凝胶电泳(A);均一化cDNA琼脂糖凝胶电泳(B)Fig.2 Agarose gel electrophoresis of dscDNA(A);Agarose gel electrophoresis of normalized ds cDNA(B)注:Marker为Trans2K Plus DNA Marker;1为双链cDNA;2为均一化cDNANote:Marker indicate Trans2K Plus DNA Marker;1 indicate ds cDNA;2 indicate normalized ds cDNA
2.3 大叶落地生根cDNA酵母文库构建与质量检测
将ds cDNA转化至提前制备好的Y187酵母感受态细胞中。菌液稀释液铺展在100 mm SD/-Leu平板上倒置培养3~4 d后,在1 000倍稀释液的平板上大约有酵母单菌落320个(图3A)。根据文库滴度和文库总容量公式可以得出所构建的文库滴度为3.2×106CFU·mL-1,文库总容量为4.8×107CFU。

图3 不同浓度文库菌液的单菌落Fig.3 The clone of different concentrations library suspension注:A为稀释1 000倍的菌液;B为稀释100倍的菌液;C为稀释10倍的菌液Note:A indicate 10-3 library dilution;B indicate 10-2 library dilution;C indicate 10-1 library dilution
从100 mm SD/-Leu平板上随机挑取24个酵母单菌落进行PCR扩增检测随机插入片段长度。电泳结果显示(图4),重组率为91.7%,24个单克隆中除第10和23无条带外其余均有清晰的单一条带,且扩增条带大小不一,插入片段长度在250~2 000 bp范围内。

图4 大叶落地生根cDNA酵母文库插入片段大小检测Fig.4 The insertion size range of yeast cDNA library of Kalanchoe daigremontiana注:Marker为Trans2K Plus DNA Marker;1~24为扩增产物Note:M indicate Trans2K Plus DNA Marker;1~24 indicate amplifications products
2.4 诱饵载体pGBKT7-KdSAHH构建及酵母转化
将已完成PCR扩增的KdSAHH片段与酶切后的pGBKT7诱饵载体纯化连接,然后将连接产物转化至DH5α大肠杆菌中。挑取单菌落摇菌,然后进行菌液PCR鉴定及琼脂糖凝胶电泳检测(图5),1 800 bp附近出现清晰条带,将该菌液送至生物公司测序。返还测序结果正确,诱饵载体pGBKT7-KdSAHH构建成功。提取测序正确的pGBKT7-KdSAHH重组质粒,并将其转化至Y2HGold酵母感受态。取100 μL菌液均匀铺展到SD/-Trp培养基上,30℃培养2~3 d。菌落能正常在营养缺陷培养基上生长,说明表达蛋白对酵母无毒害作用。
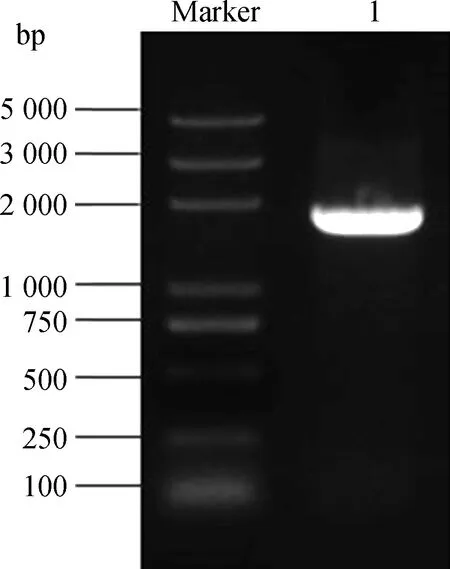
图5 诱饵载体琼脂糖凝胶电泳Fig.5 Agarose gel electrophoresis of bait vector注:Marker为Trans2K Plus DNA Marker;1为诱饵载体Note:Marker indicate Trans2K Plus DNA Marker;1 indicate bait vector
2.5 载体转录自激活检测
通过观察酵母单菌落在营养缺陷型培养基上的生长情况和生长状态进行转录自激活检测。酵母菌在SD/-Trp固体培养基上正常生长,在SD/-Trp/X-α-Gal固体培养基上正常生长且不显蓝色,在SD/-Trp/X-α-Gal/AbA固体培养基上不能正常生长且不显蓝色(图6)。载体转录自激活检测表明,KdSAHH不具备转录自激活活性,可以进行下一步酵母杂交试验筛选互作蛋白。

图6 pGBKT7-KdSAHH载体转录自激活检测Fig.6 Self-activation detection of pGBKT7-KdSAHH注:A为稀释1 000倍的菌液;B为稀释100倍的菌液;C为稀释10倍的菌液Note:A indicate 10-3 library dilution;B indicate 10-2 library dilution;C indicate 10-1 library dilution
3 讨论与结论
cDNA文库已广泛应用于生物技术领域的各项研究,因此构建高容量的cDNA文库是进行有效研究的重要基础[14]。文库质量主要从文库代表性及重组序列的完整性这两个指标进行评价[15-16]。本研究建库材料来源于大叶落地生根各个植物组织,为cDNA种类的完整性提供保障。经过文库菌液稀释涂板检测(图3A),文库滴度为3.2×106CFU·mL-1,达到低丰度mRNA筛选要求(>1×106CFU·mL-1),文库总容量为4.8×107CFU,由此可见,所建文库含有的遗传信息较丰富,文库代表性好。随机挑取24个酵母单菌落进行PCR扩增(图4),检测随机插入片段长度在250~2 000 bp范围内,文库多态性好,重组率为91.7%。同时,通过观察含有pGBKT7-KdSAHH载体的酵母菌在营养缺陷型培养基上的生长情况和生长状态,证明KdSAHH不具备转录自激活活性,可用于后续酵母双杂交试验。
本研究采用由美国Clontech公司推出的SMART技术[17]和DSN技术[18]构建大叶落地生根全长cDNA文库。将SMART技术所需RNA少、操作步骤少、用时较短且获得的文库全长比例高等优点与DSN酶的选择性降解双链核酸分子中的DNA这一特点结合起来,解决了建库过程中mRNA纯化效果不好且低丰度表达的基因易丢失的问题[19],同时还增加筛到稀有基因的可能性。但用上述方法构建的文库中有个别克隆片段偏小,插入片段的平均长度略小于1 000 bp。韦春艳等[20]在构建陆地棉酵母双杂交cDNA文库时,文库插入片段同样偏小,但并未影响后续试验的开展。总体来说,本试验构建的大叶落地生根cDNA酵母文库质量较高,满足后续酵母杂交试验的要求。
目前,在大叶落地生根不定芽发生过程中已经鉴定出一些重要调控蛋白[21-22],但具体的信号转导途径与分子调控网络尚不清楚。以已知调控蛋白为诱饵,构建诱饵表达载体,通过酵母杂交技术在大叶落地生根cDNA酵母文库筛选互作蛋白,该研究思路为探究大叶落地生根基因功能及功能基因的调控网络提供帮助。本研究成功构建了高容量的大叶落地生根cDNA酵母文库和pGBKT7-KdSAHH诱饵载体,转录自激活检测表明KdSAHH无转录自激活活性。接下来利用酵母杂交技术进行KdSAHH互作验证等试验的开展将为揭示KdSAHH基因调控下游响应基因从而影响不定芽发生的分子机制奠定研究基础。
