苯丙氨酸羟化酶基因突变检测对苯丙酮尿症的诊断意义
2021-11-08韩宗兰王兰英王海楠
韩宗兰,王兰英,王海楠
苯丙酮尿症(phenylketonuria,PKU)作为儿科临床诊治过程中的一种罕见疾病之一,其发生与机体苯丙氨酸(PA)代谢途中缺乏苯丙氨酸羟化酶(PAH)而使其不能转变成酪氨酸有关,和酮酸(keto acid,KA)蓄积也息息相关[1],属常见的氨基酸代谢病之一。国家卫生健康委员会等5部门已于2018年5月11日将其收入《第一批罕见病目录》[2]中。PKU的临床表现不均一,病人常表现为智力低下、精神神经异常及湿疹、皮肤抓痕征等。这不仅严重影响了病人的生命安全与生活质量,也增加了社会负担。据统计[3],中国的PKU发病率约0.01%,北方患病率显著高于南方,可见该病有显著的地域性特征。相关研究[4]证实,多数PKU病人是由于苯丙氨酸羟化酶基因突变而导致机体细胞内的PAH缺乏或功能下降,继而迫使PA在血液中呈高表达,而在其代谢途中损害脑及神经细胞。既往研究[5]表明PAH突变检测对增强PKU筛选水平有重要意义,借此,本文将选取135例PKU患儿进行临床实验,探讨PAH突变检测在PKU诊断中的价值和对PKU患儿的影响。现作报道。
1 资料与方法
1.1 一般资料 按中华人民共和国卫生部制定的《苯丙酮尿症和先天性甲状腺功能减低症诊治技术规范[卫妇社发(96)号][6]本院2013年1月至2019年1月收治的PKU患儿135例进行回顾性实验,纳入标准:(1)符合上述纳入标准者;(2)临床检测时血清PAH浓度≥360 μmol/L者;(3)年龄≤12岁者;(4)除PKU外无其他疾病者;(5)家属签署《知情同意书》和自愿参与本次实验及临床资料完整者。排除标准:(1)治疗期间死亡,放弃治疗及失访者;(2)合并患先天性、神经性、器质性疾病者。
1.2 方法
1.2.1 PKU筛查、诊断及分型 (1)全血DNA提取:所有受试者入院后均经本院检验科专业人员抽取PKU患儿的适量肘静脉血并提取(血卡快速酚/三氯甲烷抽提法)其全血DNA(N P-968型核酸自动提取仪,生产单位:西安天隆科技有限公司),具体的全血DNA操作步骤均严格按照Ex-DNA全血二代DNA提取试剂盒(西安天隆科技有限公司提供)相关步骤执行。(2)诊断[7]:轻度PKU:360 μmol/L<血PAH浓度<1 200 μmol/L,经典型PKU:血PAH浓度≥1 200 μmol/L。(3)基因分型:本次实验中的PAH基因分型均采用SNaPShot基因分型技术(南京医科大学附属苏州医院生殖与遗传中心实验室建立)进行分型检测,具体操作步骤均、反应条件均按文献[8]报道执行。具体操作:一是获取基因后按实验操作进行聚合酶链式反应(polymerase chain reaction,PCR)技术处理并采用多重连接探针扩增技术(multiplex ligation-dependent probe amplification,MLPA)扩增与提纯;二是对扩增后的标本进行SNaPShot延伸标志反应处理后再次提纯,最后对标本实施毛细管电泳处理。而对SNaPShot分型处理不能准确获取的PAH基因(患儿及其父亲、母亲)则再次采用其他(NGS技术)技术进行处理。(4)Sanger测序验证:将上述步骤操作,尤其是SNaPShot和NGS分型处理后的PAH基因分型纳入Sanger测序验证。
1.2.2 治疗与随访 (1)治疗:本次所纳入的受试者均结合其PAH浓度科学、合理的补助低PHE特殊食品低苯丙氨酸(2个月以内需50~70 mg·kg-1·d-1,3~6个月约40 mg·kg-1·d-1,2岁为25~30 mg·kg-1·d-1,4岁以上10~30 mg·kg-1·d-1,能维持血中苯丙氨酸浓度在0.12~0.6 mmol/L(2~10 mg/dL)为宜,持续干预1年食品喂养的基础上,辅以母乳和牛奶干预。所有受试者诊断时间均为新生儿喂奶3 d后或入院后采集其足跟末梢血进行检测,确诊之日起开始进行饮食或药物干预,干预时间。(2)随访:随访1年,1年后由儿童保健科的3名医生负责,并做好各项数据记录、整理、分析与存档。
1.3 评价标准 所有患儿的智商(intelligence quotient,IQ)均采用中文版的《学龄前期智力量表》[9]对PKU患儿的言语智商(verbal intelligence quotient,VIQ)、操作智商(performance intelligence quotient,PIQ)及全智商(full intelligence quotient,FIQ)进行评定,即将Z的0分增值至100分,Z=15(每个Z的增值),其中离差智商=100+15Z,上述三项指标中每项满分100分,得分越高智商越好。PA、IQ检测时间包括入院确诊后和持续干预1年后两个时间点。
1.4 统计学方法 采用配对t检验和Pearson相关分析。
2 结果
2.1 SNaPShot基因分型检测及Sanger测序验证与检测效果 经检测发现,本文所纳入的135例PKU患儿(270对PAH等位基因)中,男70例,女65例,年龄0~10岁,治疗时长0.5~1.0年,所有患儿基线资料与基础治疗方案无明显差异(P>0.05),其中共有12例患儿有PAH基因突变,占比8.89%,突变位点共12个,其中R243Q、Y204C、R241C、IVS4-1、R413P、R111X、R261Q、W326X及Y356X等均有突变频率,各占总PAH等位基因的5.19%、5.19%、3.70%、2.96%、2.22%、1.48%和1.48%、0.74%、0.74%,而E56D、F161S、A345T等则不发生突变(见表1)。
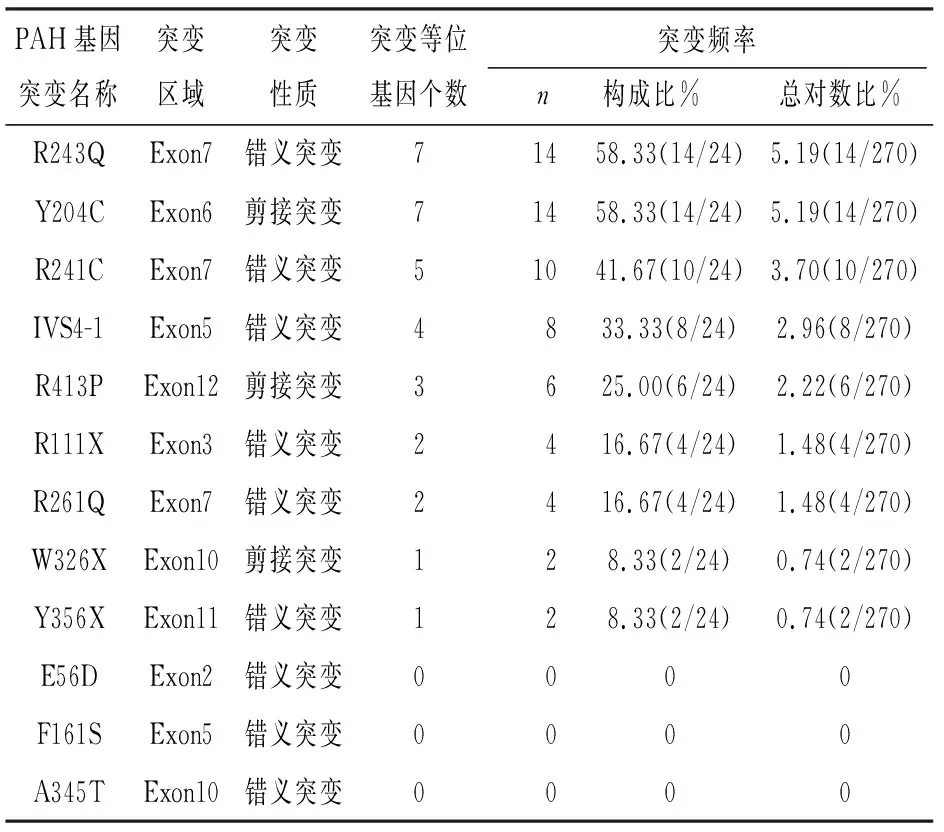
表1 SNaPShot检测及Sanger测序对12例PAH基因突变的PKU患儿在国内常见12个突变位点基因型分布检测结果
2.2 12例PAH基因突变患儿的家庭的家系基因诊断结果 经SNaPShot、Sanger及MLPA检测12例PKU患儿的PAH基因突变情况来看,纯合突变2例,占比16.67%(2/12),而复合、杂合突变10例,占比83.33%(10/12)(见表2)。
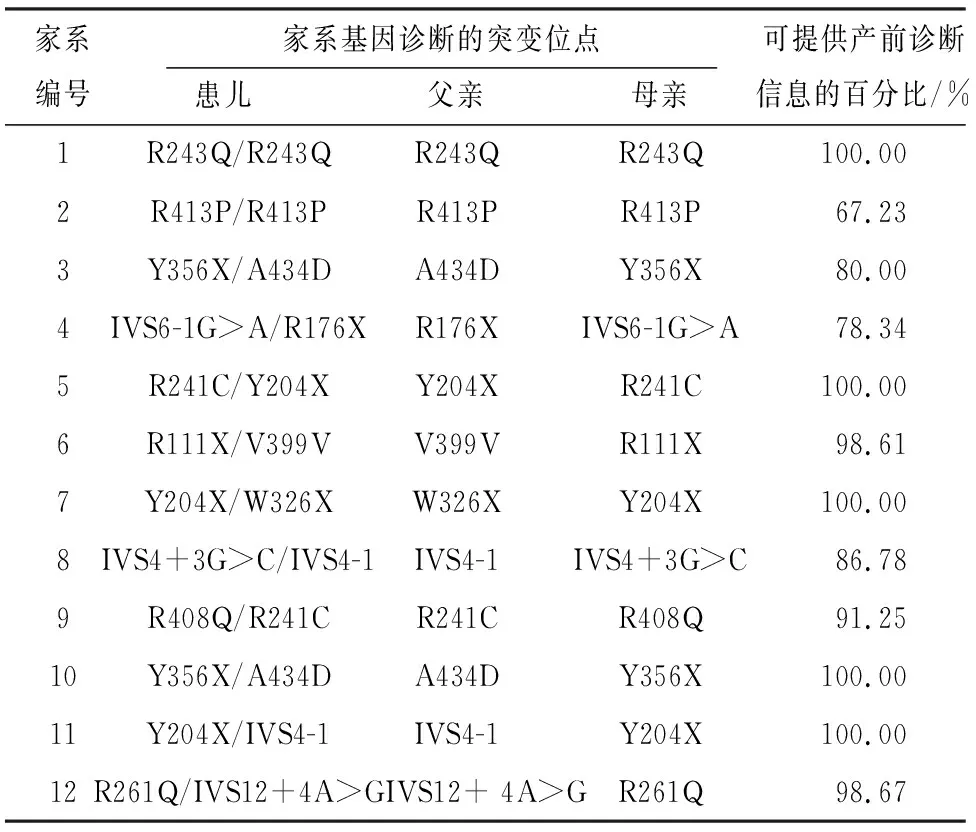
表2 SNaPShot、Sanger及MLPA检测对12例PKU患儿的家系基因诊断情况统计表
2.3 12例PAH基因突变的PKU患儿临床干预前后的血清PA水平及IQ测定 12例PKU患儿干预后的PA低于干预前、FIQ评分高于干预前,差异均有统计学意义(P<0.05),而VIQ、PIQ干预前后差异均无统计学意义(P>0.05)(见表3)。
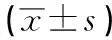
表3 12例PAH基因突变的PKU患儿临床干预前后的血清PA水平及IQ测定
2.4 12例PAH基因突变的PKU患儿的PHE平均控制浓度与1周岁时的IQ检测值 经Pearson分析发现,12例PAH基因突变的PKU患儿治疗期的PAH平均控制浓度与1周岁时的IQ检测值呈负相关(r=-0.924,P<0.01)(见表4)。
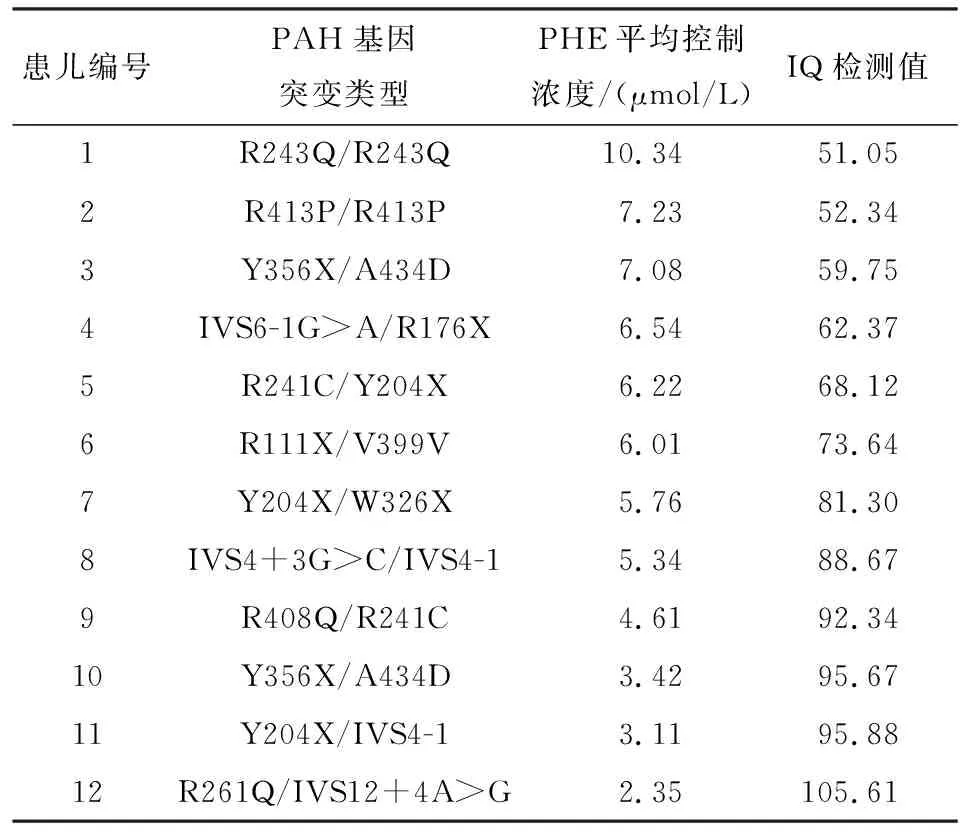
表4 12例PAH基因突变的PKU患儿的PAH平均控制浓度与1周岁时的IQ检测值相关性分析
3 讨论
PKU的发病人群较为广泛,成人、儿童乃至婴幼儿都可能患病。既往研究[10~11]证实,PKU预后效果的好坏与饮食干预、治疗依从性、PAH平均控制浓度等均有极大关系。对于PKU患儿而言,其神经系统病变的发生多在其出生后1个月左右逐渐出现。正因如此,临床上依旧将早发现、早诊断、早治疗作为预防和治疗该病的基本原则。有学者[12]通过新生儿疾病筛查诊断并实施早期治疗的大多数儿科疾病病人中,虽然大部分患儿的体质量指数(BMI)、体格及智力发育与正常儿童均相对接近,但PKU患儿的IQ与其他儿科疾病患儿的IQ相比却相对较低。PKU发展及预后均有可能影响PKU患儿的智力发育,而PAH基因突变作为导致PKU的主要病因之一,对其进行检测无疑十分必要。
据人类基因突变数据库纳入的相关数据显示,截至2017年4月,该数据库中收录的PAH基因中有突变点的基因就达610种,在这些突变基因中相对常见的是错义突变,而占比最大的应属外显子7突变,占比约16.0%。而结合目前国内研究结果表明,我国的PAH基因突变类型中,第6、7、12号外显子极常见与多发[13]。而朱海燕等[14]的研究则证实,在这些PAH突变基因中,其突变率由高到低的分别是R243Q、IVS5-96A>G、R111X等,除此之外,包括外显子7在内的基因突变情况也相对集中。由此可见,PAH基因突变是导致PKU的主要致病因素已成为不争的事实。近年来,虽然临床医学在PAH基因突变研究方面已取得较好的临床研究成绩,但其突变研究大多集中于PAH基因编码序列、内含子或内含子与外显子交界区,而忽视了对PAH基因调节区的研究。如ROSS等[15]指出,肝细胞核因子-1(hepatic nuclear factor-1,HNF-1)作为机体中的一种重要的转录因子,其在PAH基因表达调控过程中扮演重要角色,值得注意的是,HNF-1与PAH基因的结合位点主要集中于PAH基因5′侧翼。而有学者[16]也在其研究中发现,HNF-1在PAH基因表达中存在时间的长短及所扮演的促进PAH基因表达角色的好坏还与肝细胞二聚化因子(Dimerization cofactor of hepatocyte,DcoH)密切相关。当然近年来的研究再次证实PAH基因5′调控去中有3 700 bp缺失迹象[17]。以上研究均说明,PKU的发生不仅与PAH基因突变有关,也引起PAH基因调节区发生突变。
本文中,通过对135例PKU患儿的研究后发现,发生PAH基因突变者共12例(8.89%),突变位点共12个,其中R243Q、Y204C、R241C、IVS4-1、R413P、R111X、R261Q、W326X及Y356X等9个突变点的突变频率各占总PAH等位基因的5.19%、5.19%、3.70%、2.96%、2.22%、1.48%、1.48%、0.74%、0.74%,而E56D、F161S、A345T等后三个突变点的突变频率在本文中则不显著,这与王本敬等[18]的研究结果相近。而本文中的SNaPShot、Sanger及MLPA检测结果也发现,在12例PKU患儿的PAH基因突变中纯合突变率仅16.67%,复合、杂合突变则高达83.33%。而经过家系基因诊断来看,这12例PAH基因突变患儿的上述基因突变基本上均来自父母亲双方,而单纯来源于父亲或母亲的概率则极低。文献[19]研究发现,在SNaPShot检测中的上述12种PAH基因突变类型,无论是纯合突变,还是其他(复合、杂合)突变都是导致PKU的主要致病因素。这再次证实PAH基因突变是导致PKU的主要因素。那么PAH基因突变与PKU患儿的智力发育是否有关呢?许静等[20]研究发现,PAH水平在干预后与PKU患儿的VIQ、PIQ、FIQ均无显著相关性,但PAH的波动情况与患儿的FIQ则呈负相关(r=-0.78,P<0.05),而本文中的Pearson分析发现,随着PAH平均控制浓度的逐渐降低,IQ检测值则逐渐递增,PKU患儿治疗期的PHE平均控制浓度与1周岁时的IQ呈负相关(P<0.05)。本文与上述研究均有相似之处,当然也有不同之处,这在于许静等未对PKU患儿的预后情况进行跟踪随访,而本文则对12例PAH基因突变患儿进行了为期1年的随访。同时在本文中还发现,12例PKU患儿干预后的PA低于干预前、FIQ评分高于干预前(P<0.05),而VIQ、PIQ干预前后均无显著差异(P>0.05)。可见结合PKU患儿PAH基因突变情况科学、合理、严格的控制PAH血浓度对提升PKU患儿的IQ有一定的促进效能。而吴志君[21]研究15例PKU患儿后发现,在对血PAH平均浓度行科学、合理的控制后,80.0%(12/15)PKU患儿的IQ均有所改善,且有33.3%(5/15)的PKU患儿的IQ改善与健康儿童相比无差异。而在该研究中,当将血PHE平均浓度控制在(3.26±1.64)mg/dL时,PKU患儿整体的IQ值≥90。综上,在准确掌握PAH基因突变的前提下,科学、合理地控制PAH水平对改善PKU患儿的IQ有一定的帮助。PAH基因突变与IQ的相互作用主要体现在以下几个方面:一是PA在转化为酪氨酸的过程中除了需要PAH外,还必须要有辅酶四氢生物蝶呤(Tetrahydrobiopterin,BH4)的参与,而当PAH发生基因突变时,则会导致包括BH4在内的相关酶的活性缺陷,最终导致PA无法正常转化成酪氨酸而致使血液、脑脊液及相关组织中堆积PA;二是PA代谢为酪氨酸的通路受阻,相关的旁路代谢则会增强[21],而当PA经肝脏代谢时会产生大量的有害代谢物,继而导致脑损伤。三是PKU的典型症状是生长发育和精神发育迟缓。临床实验[22]表明,随着辅食剂量的不断添加,对PKU患儿的智力发育越不利,而智力发育落后又会随着大脑的逐渐成熟而日趋稳定,其根源可能与父母双方是致病基因变异的携带者有关。但更深层的研究仍有待更多的研究数据佐证。
综上所述,在PKU病人的临床治疗过程中科学、合理地进行PAH基因突变检测并严格控制患儿的PAH血浓度有较好的临床应用与推广价值,也是改善患儿IQ的关键因素之一,家长、医学界和社会各界应高度重视。但就本研究情况来看,虽然在探析PAH基因突变的类型方面做了对照分析,对PAH基因突变与PKU患儿IQ的关系进行了比较研究,而证实了PAH基因突变对患儿的IQ水平指标也存在一定的影响,但对其他水平的影响还待进一步研究,故期待在未来的日子里有机会予以弥补。
