Acute liver failure with hemolytic anemia in children with Wilson’s disease: Genotype-phenotype correlations?
2021-11-04TudorLucianPopAlinaGramaAnaCristinaStefanescuClaudiaWillheimPeterFerenci
Tudor Lucian Pop, Alina Grama, Ana Cristina Stefanescu, Claudia Willheim, Peter Ferenci
Tudor Lucian Pop, Alina Grama, Ana Cristina Stefanescu, 2nd Pediatric Discipline, Department of Mother and Child, Iuliu Hatieganu University of Medicine and Pharmacy, Cluj-Napoca 400177, Romania
Tudor Lucian Pop, Alina Grama, 2nd Pediatric Clinic, Center of Expertise in Pediatric Liver Rare Disorders, Emergency Clinical Hospital for Children, Cluj-Napoca 400177, Romania
Claudia Willheim, Peter Ferenci, Department of Internal Medicine III, Gastroenterology and Hepatology, Medical University of Vienna, Wien A-1090, Austria
Abstract BACKGROUND Wilson’s disease (WD) is a rare autosomal recessive inherited disorder of copper metabolism.Acute liver failure (ALF) and hemolytic anemia represent the most severe presentation of WD in children.No clear genotype-phenotype correlations exist in WD.Protein-truncating nonsense, frame-shift, or splice-site variants may be associated with more severe disease.In contrast, missense variants may be associated with late-onset, less severe disease, and more neurological manifestations.Recently, a gene variant (HSD17B13:TA, rs72613567) with a possible hepatic protective role against toxins was associated with a less severe hepatic phenotype in WD.AIM To analyze the possible genotype-phenotype correlations in children with WD presented with ALF and non-immune hemolytic anemia.METHODS The medical records of children with WD diagnosed and treated in our hospital from January 2006 to December 2020 were retrospectively analyzed.The clinical manifestations (ALF with non-immune hemolytic anemia or other less severe forms), laboratory parameters, copper metabolism, ATP7B variants, and the HSD17B13:TA (rs72613567) variant were reviewed to analyze the possible genotype-phenotype correlations.RESULTS We analyzed the data of 51 patients with WD, 26 females (50.98%), with the mean age at the diagnosis of 12.36 ± 3.74 years.ALF and Coombs-negative hemolytic anemia was present in 8 children (15.67%), all adolescent girls.The Kayser-Fleisher ring was present in 9 children (17.65%).The most frequent variants of the ATP7B gene were p.His1069Gln (c.3207A>G) in 38.24% of all alleles, p.Gly1341Asp (c.4021G>A) in 26.47%, p.Trp939Cys (c.2817G>T) in 9.80%, and p.Lys844Ter (c.2530A>T) in 4.90%.In ALF with hemolytic anemia, p.Trp939Cys (c.2817G>T) and p.Lys844Ter (c.2530A>T) variants were more frequent than in other less severe forms, in which p.His1069Gln (c.3207A>G) was more frequent.p.Gly1341Asp (c.4021G>A) has a similar frequency in all hepatic forms.For 33 of the patients, the HSD17B13 genotype was evaluated.The overall HSD17B13:TA allele frequency was 24.24%.Its frequency was higher in patients with less severe liver disease (26.92%) than those with ALF and hemolytic anemia (14.28%).CONCLUSION It remains challenging to prove a genotype-phenotype correlation in WD patients.In children with ALF and hemolytic anemia, the missense variants other than p.His1069Gln (c.3207A>G) and frame-shift variants were the most frequently present in homozygous status or compound heterozygous status with site splice variants.As genetic analysis is usually time-consuming and the results are late, the importance at the onset of the ALF is questionable.If variants proved to be associated with severe forms are found in the pre-symptomatic phase of the disease, this could be essential to predict a possible severe evolution.
Key Words: Wilson’s disease; Children; Acute liver failure; Hemolytic anemia; ATP7B variant; Genotype-phenotype correlation
INTRODUCTION
Wilson’s disease (WD) is a rare autosomal recessive inherited disorder of copper metabolism caused by homozygous or compound heterozygous variants of theATP7Bgene.The prevalence of WD is estimated as 1/30000[1].TheATP7Bgene encodes transmembrane copper-transporting ATPase (ATP7B) and is located on chromosome 13q14.3, containing 20 introns and 21 exons[2].According to the Human Gene Mutation Database, more than 800 variants of theATP7Bgene have been described.More than half of these variants are single nucleotide missense and nonsense, and the others are insertions/deletions and splice-site variants[3,4].
The clinical forms of WD are very variable due to the copper accumulation in different organs.The age of onset has been reported to be between 2 and 70 years[5,6].Liver disease is the first clinical manifestation in 40%-60% of WD patients, more often in the first decade[1,5,7,8].In children, WD patients present with an incidental finding of high levels of transaminases in an asymptomatic child, acute or chronic hepatitis, or decompensation of cirrhosis (in older children and adolescents)[5,9].As an initial form of presentation, neurological disease is described in 18%-68% of patients, mainly in young adults (20-30 years).WD’s most common neurological features are tremor, dystonia, parkinsonism, associated with dysarthria, gait and posture disturbances, drooling, and dysphagia[1].Also, psychiatric disease (mainly mood disturbances, depression, or bipolar disease) may be present, mainly in adulthood.A decline in school performances, impulsiveness, and inappropriate behavior was reported in adolescents[1].Hematologic disease, renal disease, skeletal and cardiac disease may be described in WD patients[1,10,11].
Acute liver failure (ALF) may be the initial presentation or a complication during WD evolution in children and young adults[12].Approximately 2%-6% of ALF cases may be caused by WD[13,14].A rare presentation of WD (5%[15]), ALF accompanied by a hemolytic crisis may have a severe evolution, with coagulopathy, encephalopathy, and progressive renal failure, resulting in death without an emergency liver transplantation[1,10,11,16].This clinical form of WD occurred in 30% of children with ALF requesting liver transplantation and 60% of those with fatal evolution before transplantation[5].Therefore, early diagnosis and referral to specialized centers are determinants for the prognosis in these patients[12].
There is a continuous interest in genotype-phenotype correlations in WD.Based on the phenotypic classification, studies tried to find a link between the genetic variants and clinical forms or severity of WD disease, important for the prognosis of the disease[2,17].TheATP7Bgene variants may have different effects on the presence and function of the ATPase encoded with various consequences on the clinical presentation.Many studies have tried to analyze these correlations regarding the age of the onset, neurological or hepatic form, ceruloplasmin activity, hepatic copper level, or the presence of Kayser-Fleischer (KF) ring[18-20].Still, there is no definite genotypephenotype correlation so far, which may be due to the disease’s high genetic heterogeneity and rareness[2,21].Some authors suggest that the severe hepatic phenotype and earlier onset are more likely associated with the nonsense or frame-shift variants.A less severe hepatic or a neurologic phenotype is linked to missense variants [2,22-26].The clinical presentation in WD may also be influenced by environmental and epigenetic factors or modifier genes[4].Recently, a gene variant (HSD17B13:TA, rs72613567) with a possible hepatic protective role against toxins was associated with a less severe hepatic phenotype in WD[27].
Our study aimed to analyze the possible genotype-phenotype correlations in children with WD presented with ALF and non-immune hemolytic anemia and to investigate the most commonATP7Bvariants in our patients with this severe form of the disease.
MATERIALS AND METHODS
The medical records of children with WD diagnosed and treated in our hospital from January 2006 to December 2020 were retrospectively analyzed.The clinical manifestations (acute or chronic liver disease, neurologic disease, ALF with non-immune hemolytic anemia), laboratory parameters, copper metabolism, andATP7Bvariants were reviewed.
Diagnostic of WD was based on positive family history, clinical symptoms (including the presence of KF ring), and laboratory tests (low serum ceruloplasmin, < 20 mg/dL, elevated 24-h urinary copper excretion, baseline or stimulated by penicillamine) following the current diagnostic and management guidelines[28,29].ALF with hemolytic anemia was diagnosed on the coagulopathy (prolonged prothrombin time, increased international normalized ratio (INR) > 2 without hepatic encephalopathy or > 1.5 in the presence of encephalopathy), low hemoglobin level, and negative Coombs test.Laboratory tests were performed using standard methods.None of our patients had a liver biopsy to assess the histology, as we could not measure the copper content in our service.The severity of the fibrosis was evaluated at diagnosis and during the follow-up using a non-invasive assessment of liver stiffness by transient elastography (FibroScan, Echosense, France)[9].
The molecular analysis of theATP7Bgene was performed using a semi-nested polymerase chain reaction-based restriction fragment length polymorphism assay for p.His1069Gln (c.3207A>G) variant detection as previously described.If negative or heterozygous for p.His1069Gln (c.3207A>G) variant, samples were Sanger sequenced by the ABI Prism 310 Genetic Analyzer (Perkin Elmer; Norwalk, CT, United States) until 2012, followed by the 3500 Genetic Analyzer (Applied Biosystems; Foster City, CA, United States) using published primers.
The HSD17B13:TA (rs72613567) variant was determined using allelic discrimination real-time polymerase chain reaction and validated by Sanger sequencing in normal controls having different HSD17B13 genotypes.Unfortunately, this evaluation was technically possible only for the second half of the study, and only 33 patients were assessed.We included only the children with WD confirmed by molecular analysis, and we excluded all suspected WD patients without genetic confirmation or with incomplete data.
We analyzed the clinical and laboratory features, including the most frequent variants in children with ALF and hemolytic anemia compared to those with other clinical forms.
Statistical analysis of the data collected was performed using Statistica 13.5 (Tibco Software; Palo Alto, CA, United States).The variables with normal distribution were presented as mean and standard.Comparison of continuous variables was performed using the Studentt-test.Categorical variables were presented as numbers and percentages; they were compared using the Chi-square test.Two-sidedPvalues were analyzed, and thePvalue < 0.05 was considered statistically significant.
RESULTS
During the last 15 years, 67 patients with WD were diagnosed and treated in our clinic.After reviewing genetic data, we included 51 patients, 26 females (50.98%), with the mean age at WD diagnosis of 12.36 ± 3.74 years (between 5 and 23 years).
Almost all patients included in our study presented liver diseases; only one was with a neurological form, and one was diagnosed following the screening due to WD in the family.Our clinic is the main pediatric hepatology service and center for expertise in pediatric liver rare disorders in Transylvania, Romania.Therefore, the selection of the patients referred to our center would be biased regarding the clinical presentation in our WD patients.ALF and Coombs-negative hemolytic anemia was present in 8 children (15.67%), all adolescent girls.The KF ring was present in 9 children (17.65%).The clinical and laboratory characteristics of the WD patients included in this study, based on the clinical onset, are presented in Table 1.
In two girls, ALF with hemolytic anemia was not the initial presentation that led to the WD diagnostic.Initially, they had only increased transaminases but progressed shortly to this severe clinical evolution.
In our patients, the most frequent variant of theATP7Bgene was p.His1069Gln (c.3207A>G), present in 12 children in homozygous status and 17 children in compound heterozygous status (38.24% of all alleles).p.Gly1341Asp (c.4021G>A) variant was present in 10 children in homozygous status and 7 patients in compound heterozygous status (26.47%).The other two frequent variants (mainly in patients with ALF and hemolytic anemia) were p.Trp939Cys (c.2817G>T), in 5 children in homozygous status (9.80%) and p.Lys844Ter (c.2530A>T), in one child as homozygous status and three children as a part of compound heterozygous status (4.90%).In Table 2, we present the most frequent variants grouped by the clinical form of presentation.In ALF with hemolytic anemia, p.Trp939Cys (c.2817G>T) and p.Lys844Ter (c.2530A>T) were more frequent than in other less severe forms, in which p.His1069Gln (c.3207A>G) was more frequent.p.Gly1341Asp (c.4021G>A) variant has a similar frequency in all hepatic forms.
For 33 of the patients included in our study, the HSD17B13 genotype was evaluated.The overall HSD17B13:TA allele frequency in our study was 24.24%.HSD17B13:TA allele frequency was higher in patients with less severe liver disease (26.92%) than ALF and hemolytic anemia (14.28%).Table 3 presents the demographic, clinical, and genotype association in patients investigated for the HSD17B13:TA variant.
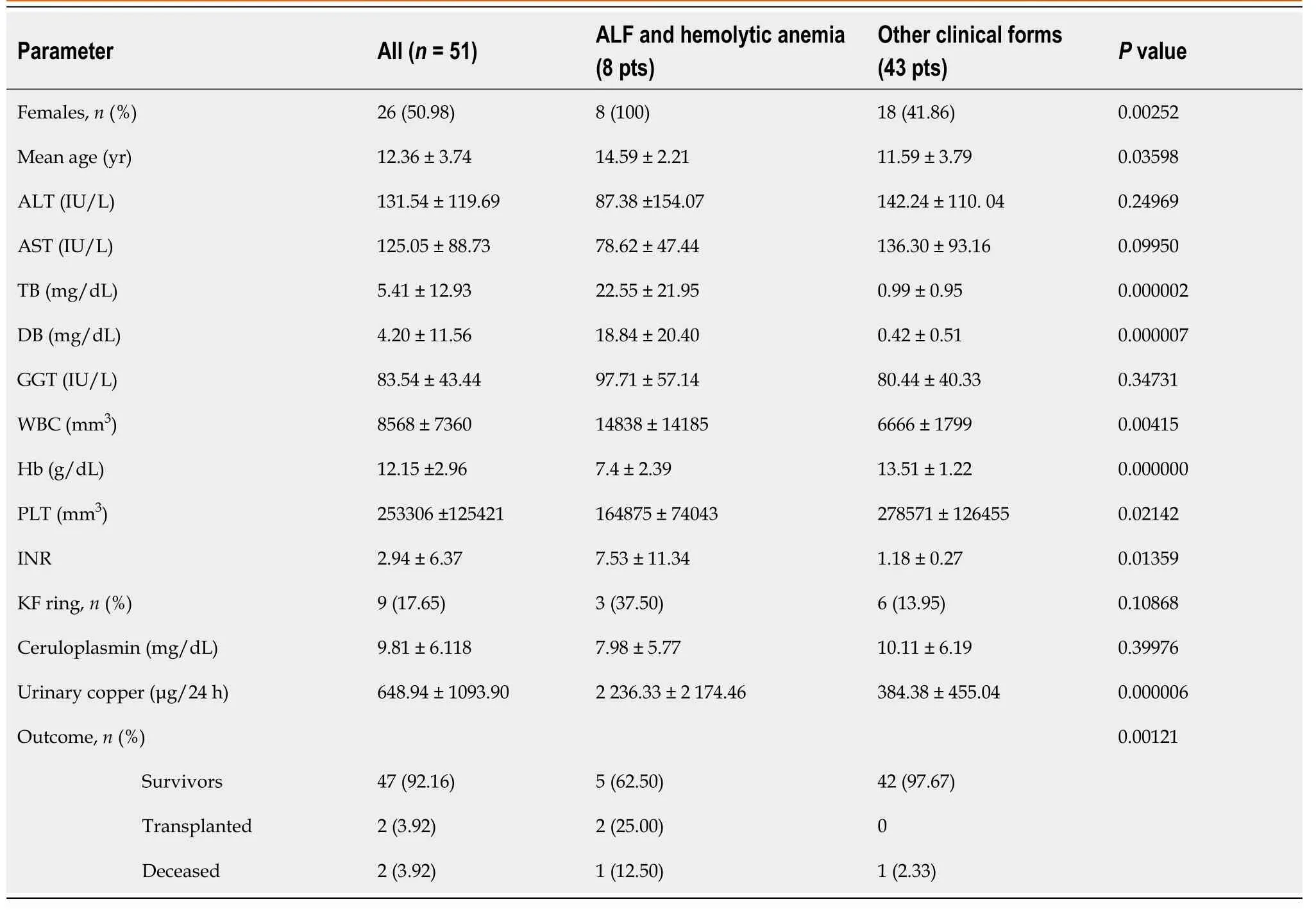
Table 1 Clinical and laboratory characteristics in Wilson’s disease children with acute liver failure and hemolytic anemia
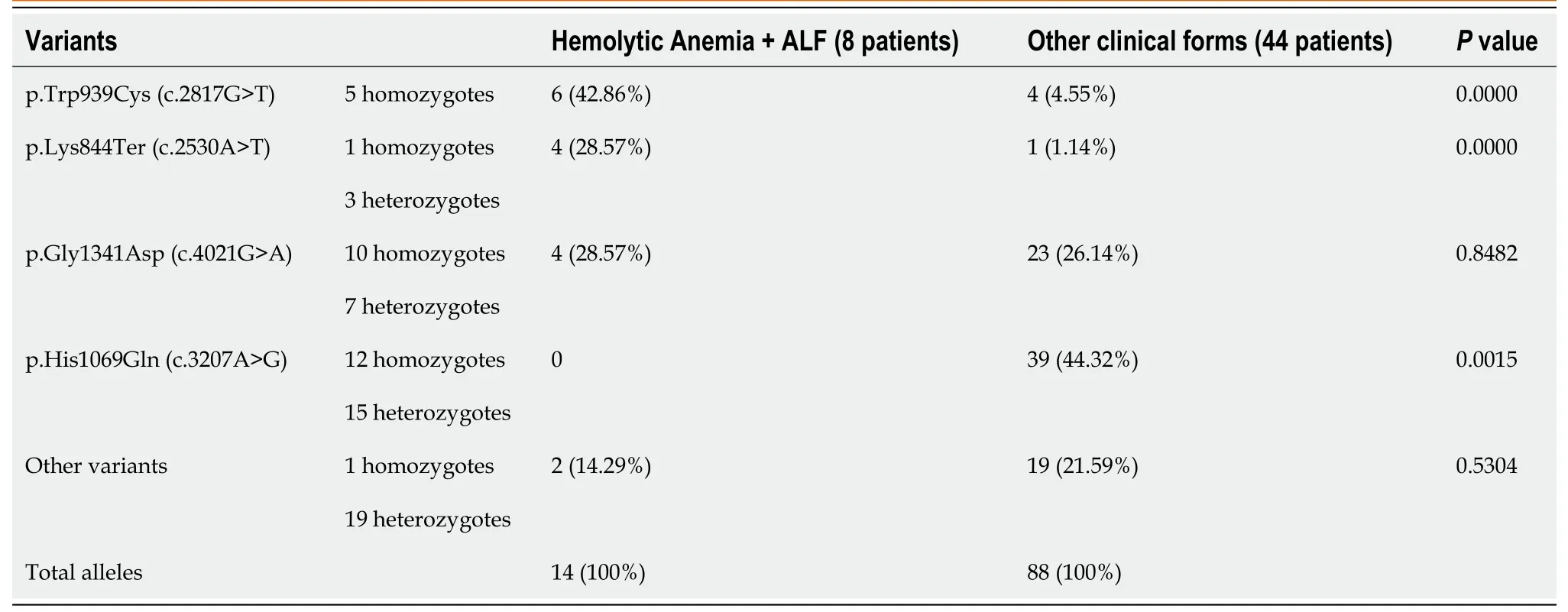
Table 2 Variants of ATP7B gene in children with hemolytic anemia and acute liver failure
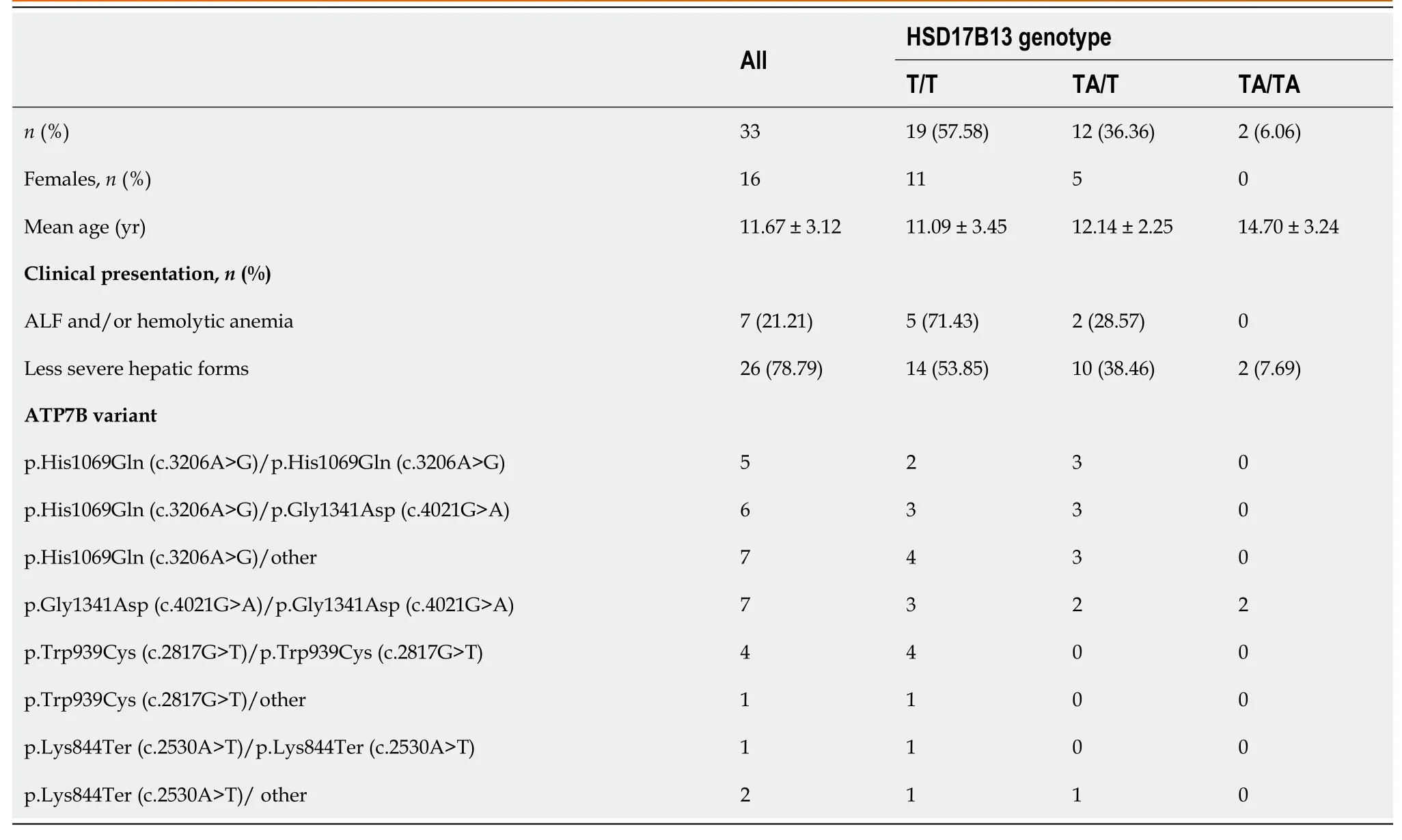
Table 3 Data regarding the patients evaluated for the HSD17B13:TA variant
Two patients with ALF were transplanted, five survived with the native liver following supportive intensive care, and one girl had a fatal evolution on the second day after admission.Also, another child with cirrhosis died due to severe complications before liver transplantation was possible.
DISCUSSION
Our study aimed to assess the possible genotype-phenotype correlations regarding WD’s most severe clinical form in children and adolescents.This endeavor in patients with WD is challenging, as was proved by many studies already published.So far, the research failed to conclude this issue due to the high heterogeneity ofATP7Bvariants(more than 1300 described) and the rareness of the disease (small series of patients).Furthermore, the increased number of compound heterozygotes involving different kinds of variants makes this analysis more difficult[18,19,24,30,31].As phenotypic differences were reported in siblings with the same genotype or monozygotic twins, the involvement of other factors is possible[2,30,32-35].Environmental factors (nutritional copper intake, infections, drugs, or other toxins), modifier genes, and epigenetic factors’ interaction with the genetic variants may explain the different clinical presentations in WD[4,8,18,22].
The introduction of a phenotype classification tried to ease analyzing the clinical forms in WD[17].Our patients mainly have the hepatic form (the most frequent one in children and adolescents).Some of our older patients also had neurological and psychiatric manifestations.Not all our patients suspected of WD had a genetic confirmation of theATP7Bvariants.Therefore, only patients with two WD variants in cis were included in our study to analyze the possible genotype-phenotype correlations.
ALF with hemolytic anemia was present in 8 children, all girls.The age of onset was higher than other hepatic presentations (acute or chronic hepatitis, autoimmune features, or cirrhosis, data not shown).The increased frequency of ALF described in females is not fully understood, but it may be explained by hormonal differences or the intervention of epigenetic factors (methylome and transcriptome differences)[4,13,18,19].
The KF ring was described in 9 children with liver disease (17.65%), three of them with ALF, and 6 in the other forms.Four of those six children with KF ring in other forms of liver disease presented neurological manifestations in their evolution.Other studies proved that ocular involvement is less frequent in hepatic than in neurological involvement[31].The presence of the KF ring was reported lower in children.In Greek children with WD, the KF ring was present in 48.7% of those with liver disease and 16% of those diagnosed through family screening[7], while in the Italian children, only in 8.6% of those with liver disease[8].The KF ring was present in more than half of the ALF patients, compared to 37.5% in our small series[10].
Regarding the laboratory results, the differences in children with ALF and hemolytic anemia and the other forms are expected for the bilirubin, hemoglobin, and INR.The number of white blood cells (WBC) is higher in children with ALF, and platelets are lower.There are no significant differences in the serum level of transaminases (even lower in ALF patients) and gamma-glutamyl transferase.The number of WBCs is an important risk factor as it was included in the prognostic score to predict mortality and evaluate the need for liver transplantation[5].Also, the low level of transaminases in children with the ALF form of WD is a well-known feature and would help the diagnostic.High aspartate-aminotransferase (AST) to ALT ratio and low alkaline phosphatase to serum bilirubin ratio may be used to differentiate the WD patients in ALF[13,15].In our cohort, two children with ALF had an AST/ALT ratio higher than 4 and only one alkaline phosphatase to bilirubin level ratio lower than 2.In children, the ratio between serum alkaline phosphatase and total bilirubin level may not always be helpful due to bone-derived alkaline phosphatase[5].The transaminase level was lower than in children with acute hepatitis.As the age is higher in patients presenting with ALF, the evolution of the disease without any clinical sign for years explains the severe fibrosis or cirrhosis in these patients.In a large study that aims to analyze the genotype-phenotype correlation, fulminant liver failure or hemolysis were associated with liver cirrhosis in 93.4% and 66.7% of patients, higher than in the other milder presentations of WD[19].
In our children with ALF and hemolytic anemia, the serum ceruloplasmin level was lower than in the other patients but not significantly.In the meantime, the urinary copper excretion was higher, as can be expected, due to the severe necrosis associated with ALF.
No clear genotype-phenotype correlations exist in WD.Protein-truncating nonsense, frame-shift, or splice-site variants have a significant functional and structural impact on the ATP7B protein and may be associated with more severe disease (early-onset, low ceruloplasmin level, high copper content in liver).In contrast, missense variants are associated with late-onset, less severe disease and more neurological manifestations[18,25,26,36].There are also reports of some missense variants associated with the early onset of disease with various severity in the same family[30].Previous reports proposed the association of exon 18-20 variants with hepatic and hematological onset but not with neurological disease[37].
The most frequent variants in Central Europe, p.His1069Gln (c.3207A>G), was also the most frequent one in our cohort.It was found in homozygous or heterozygous status in 38.24% of all alleles in our study, compared with 72% in Poland, 35% in Greece, and 38% in a previous study from Romania[17,21,23,26,38].This variant is more frequent in older patients with the neurological form of WD[3,7,23].In our cohort, there was no child with ALF and hemolytic anemia with p.His1069Gln (c.3207A>G) variant.This is a missense variant and is probably associated with protein misfolding, abnormal phosphorylation of the P-domain, and altered ATP binding orientation and affinity[13].R969Q, another missense variant present in our children, is almost exclusively associated with late-onset liver disease[3,23].
Another missense variant, p.Gly1341Asp (c.4021G>A), was the second most frequent one in our children.p.Gly1341Asp (c.4021G>A) is a variant of the transmembrane domain of theATP7Bgene and, in homozygous status, was proved to be associated with more severe and early onset of WD[39].This variant was associated in homozygous status with ALF and/or hemolytic anemia in two children.In one girl, hemolytic anemia developed after treatment with zinc for a chronic increase of transaminases with questionable compliance.The second girl with this genotype-phenotype association has a younger sister with the same genetic status presenting only an increase of transaminases.The most frequent variant in our patients with ALF and/or hemolytic anemia was p.Trp939Cys (c.2817G>T), described previously in early-onset hepatic disease and with a high risk for liver failure in homozygotes[24].Three adolescents (girls) with ALF presented this variant in homozygous status; the other two children (males) had the same status but did not have a severe form.The p.Lys844Ter (c.2530A>T) variant is the fourth most frequent in our cohort; it was present more in children with a severe form of WD.One girl was homozygote, and in another two girls, the variant was associated with splice-site variants in a compound heterozygous status.The p.Lys844Ter (c.2530A>T) variant is a frame-shift variant presumed to be associated with severe clinical evolution, as are also splice-site variants.It was previously described in WD patients of Hungarian origin[40] and few patients with late-onset of WD[41].
The early diagnosis of WD in children would probably prevent the evolution and sometimes the onset of the disease with a severe form.As mentioned in other studies, gender would modify the disease presentation due to different hormone balance[18,19].If we analyze the possible influence of the sex of the patients, the severe form of the disease was present in two of the four girls and none of the boys with p.Gly1341Asp (c.4021G>A) homozygous status.All children with p.Gly1341Asp (c.4021G>A) variant in compound heterozygous status associated with p.His1069Gln (c.3207A>G) variant experienced a less severe form of WD.
HSD17B13 encodes a protein involved in regulating the biosynthesis of lipids, and by its enzymatic roles, is implicated in lipid-mediated inflammation.Recently, a protein-truncation variant (HSD17B13:TA, rs72613567) was shown to have a protective role against liver toxins, including copper toxicity in WD[27].In our cohort, the allele frequency of HSD17B13:TA was similar to other results for the Caucasian population, higher in patients with less severe liver disease than those presented with ALF and hemolytic anemia.The age of diagnosis was higher in patients homozygous for this variant than in heterozygous status or without this variant.Even without statistical significance, these results suggest the possible role of the HSD17B13:TA variant in the modulation of the WD severity together with factors, including sex, age,ATP7Bvariant, and other gene variants.
ALF was fatal only in one of our cases included in this study.Two girls underwent emergency liver transplantation on the fourth day after their presentation in our service.The liver transplantation was performed at the Fundeni Institute in Bucharest, Romania.This clinical presentation should be regarded as an emergency[5,42].The patients should be referred as soon as possible to a center that could provide intensive care, including extrahepatic liver support, until liver transplantation would be possible for severe cases.Unfortunately, one girl died the second day after her admission to our center.
Strengths and limitations.This study presents the largest cohort of children with genetically confirmed WD from our country and the neighboring region.It represents the first description of the possible correlation of ALF and hemolytic anemia with p.Trp939Cys (c.2817G>T) and p.Lys844Ter (c.2530A>T) variants in Eastern European children with WD.However, there are some limitations of our study.Firstly, the small number of children with this severe form made the statistical analysis of our findings difficult.Another issue is represented by the selection of patients, as our pediatric hepatology service admits mainly children and adolescents with hepatic disease.A significant limitation was the difficulty of considering and analyzing other possible factors that would lead to an acute, severe clinical form compared to children with the same genotype [p.Gly1341Asp (c.4021G>A)].
In the future, with the onset of a National Registry for patients with WD, including the genetic analyzes, more data on WD patients from Romania would be available.In the severe clinical form of WD, the genetic background would be less critical from the point of view of immediate medical care.The result of the genetic analysis would arrive with the clinician late, after the evolution of the patient would be clear.With the recent progress in screening for WD[43], the genetic analysis in children with an early suspected disease would help predict future evolution.When nonsense, frame-shift, or splicing-site variants are identified in a pre-symptomatic period, the importance of this genotype-phenotype correlation for the prognostic is evident.
CONCLUSION
It remains challenging to prove a genotype-phenotype correlation in WD patients due to the small number of patients in the reported series and the increased genetic heterogeneity.In children with ALF and non-immune hemolytic anemia, the nonsense variants other than p.His1069Gln [as p.Trp939Cys (c.2817G>T)] and frame-shift variants [p.Lys844Ter (c.2530A>T)] were the most frequently present in homozygous status or compound heterozygous status with site splice variants.As genetic analysis is usually time-consuming and the results are late (except in the screening of the relative of an index patient), the importance for the prognosis at the onset of the ALF is questionable.However, if variants proved to be associated with severe forms are found early in the evolution of the disease, this could be essential to predict a possible severe evolution if the patients would not follow treatment.
ARTICLE HIGHLIGHTS
Research background
There is a continuous interest in genotype-phenotype correlations in Wilson’s disease(WD).
Research motivation
The aim is to study the possible genotype-phenotype correlations in children with acute liver failure (ALF) and hemolytic anemia in WD.
Research objectives
The objectives include the analysis ofATP7Bvariants in children with ALF and hemolytic anemia in WD compared to the other clinical presentations and the possible role of the HSD17B13:TA variant in the modulation of the WD severity.
Research methods
The retrospective study included 63 children with WD diagnosed and follow-up during 2006-2020.The clinical manifestations (acute or chronic liver disease, neurologic disease, ALF with non-immune hemolytic anemia), laboratory parameters, copper metabolism,ATP7Bvariants, and the HSD17B13:TA (rs72613567) variant were reviewed.
Research results
In our cohort, in children with ALF and non-immune hemolytic anemia, the nonsense variants other than p.His1069Gln (c.3206A>G), as p.Trp939Cys (c.2817G>T), and frame-shift variants, as p.Lys844Ter (c.2530A>T), were the most frequently present.The allele frequency of HSD17B13:TA was similar to other results for the Caucasian population, higher in patients with the less severe liver disease than those presented with ALF and hemolytic anemia.
Research conclusions
It remains challenging to prove a genotype-phenotype correlation in WD patients due to the small number of patients in the reported series and the increased genetic heterogeneity.When nonsense, frame-shift, or splicing-site variants are identified in a presymptomatic period, the importance of this genotype-phenotype correlation for the prognostic is evident.
Research perspectives
A more extensive study involving children and adolescents with ALF and hemolytic anemia form of WD should be provided to confirm the findings.New studies are needed to evaluate the role of protective variant, HSD17B13:TA (rs72613567), in association with other factors, in less severe forms of WD in children.
杂志排行

World Journal of Hepatology的其它文章
- Coronavirus disease 2019 in liver transplant patients: Clinical and therapeutic aspects
- Focal nodular hyperplasia associated with a giant hepatocellular adenoma: A case report and review of literature
- Clinical outcomes of patients with two small hepatocellular carcinomas
- Machine learning models for predicting non-alcoholic fatty liver disease in the general United States population: NHANES database
- Impact of biliary complications on quality of life in live-donor liver transplant recipients
- Serum zonulin levels in patients with liver cirrhosis: Prognostic implications