甲基转移酶EZH2在恶性肿瘤发生发展及治疗中的新进展*
2021-11-03朱永霞施丽红陈欣怡肖洪涛
朱永霞,施丽红,陈欣怡 ,肖洪涛
610041 成都,四川省肿瘤医院·研究所,四川省癌症防治中心,电子科技大学医学院 临床药学部(朱永霞、施丽红、肖洪涛);610041 成都,四川大学华西医院 生物治疗国家重点实验室(陈欣怡)
恶性肿瘤是严重威胁人类健康和社会发展的主要疾病之一。近年来,肿瘤的发病率在全球范围内仍然呈增长趋势[1-2]。Zeste基因增强子同源物2(enhancer ofZestehomolog 2,EZH2)是一种重要的甲基转移酶,其作为核心催化亚基与其他成员形成多梳抑制复合物2(polycomb repressive complex 2,PRC2),并通过调控转录活性进而促进肿瘤细胞的增殖、转移和耐药[3-4]。研究表明,EZH2在多种肿瘤组织中过表达或产生突变,且与肿瘤的发生发展及不良预后密切相关,可作为肿瘤治疗的有效靶点[5-6]。目前,多种类型的EZH2抑制剂已用于肿瘤的基础和临床治疗研究,并取得里程碑式的成功[5, 7]。本文将对EZH2在肿瘤发生发展中的作用、分子机制和相关抑制剂的研究新进展进行综述。
1 EZH2的概述
EZH2是果蝇zeste基因的人类同源物,其基因位于人源染色体7q35上,含有20个外显子,可编码含有746个氨基酸的蛋白[8]。EZH2具有多个功能结构域[9-10]:胚胎外胚层发育因子(embryonice ectoderm development,EED) 结合区域结构域;SUZ12结合区域:结构域1(D1)、结构域2(D2);2个SANT(SWI3-ADA2-N-CoR-TFIIIB)结构域,允许染色质重塑蛋白质与组蛋白相互作用的结构域;富含半胱氨酸的CXC结构域;甲基化催化结构域SET[Su(var)3-9, Enhancer ofzeste,Trithorax](图1)。

图1 EZH2的功能域示意图及SET结构域催化蛋白质上的赖氨酸残基甲基化的机制
EZH2是表观遗传抑制因子PcG蛋白家族的重要成员之一,可以与其他家族成员形成组蛋白甲基转移酶复合物PRC2 ,在SET催化结构域构成酶的催化活性中心[3, 11]。如图1和图2所示,含有SET结构域的EZH2可以催化PRC2复合物对底物蛋白质上的赖氨酸残基进行甲基化修饰,其中底物包括组蛋白H3和非组蛋白,进而参与大量基因表达的调控[12]。目前,EZH2的异常过表达与激活性突变在多种类型的癌症或肿瘤组织中被检测到,包括乳腺癌、肺癌、黑色素瘤、卵巢癌以及弥漫性大B细胞淋巴瘤(diffuse large B-cell lymphoma,DLBCL)[4, 13-15]。EZH2的异常过表达与肿瘤的转移、恶性程度及不良预后密切相关[4, 16],因此,EZH2是治疗肿瘤的有效靶点之一。
2 EZH2的作用机制
2.1 经典的PRC2依赖的H3K27甲基化的转录调控
越来越多的证据表明,EZH2可以通过多种方式发挥转录活性调控作用,其中,最早发现的是经典的PRC2依赖的H3K27甲基化的转录抑制调控[3]。如图2所示,EZH2首先与 EED、果蝇Zeste基因抑制因子(spressor ofzeste12,SUZ12)及视网膜母细胞瘤抑制因子相关蛋白(retinoblastoma suppressor associated protein 46/48,RbAp46/48)等聚合形成PRC2复合物,然后通过SET结构域催化细胞核中组蛋白H3中27位的赖氨酸三甲基化(H3K27me3),随后抑制下游靶基因的转录,进而参与调控多种基因的表达[3,17]。

图2 EZH2的作用机制图
2.2 PRC2依赖的非组蛋白甲基化的转录调控
EZH2还可以通过PRC2复合物依赖性的方式甲基化非组蛋白[18]。EZH2通过形成PRC2复合物,对心肌转录因子4(GATA binding factor 4,GATA4)中第299位的赖氨酸甲基化,从而减弱其与组蛋白乙酰转移酶p300的相互作用和乙酰化活性,促进GATA4的转录抑制和基因沉默[19]。
2.3 不依赖于PRC2的非组蛋白甲基化的转录调控
研究发现,EZH2也可以通过不依赖于PRC2的方式直接甲基化非组蛋白,进而影响下游靶基因的转录调控[18]。这种不依赖于PRC2的非经典作用方式首次报道于去势抵抗性前列腺癌(castration-resistant prostate cancer,CRPC)[20]。Xu等[20]发现,AKT通路会引起EZH2中第21位的丝氨酸发生磷酸化,磷酸化的EZH2会与雄激素受体(androgen receptor,AR)结合,引起AR及其相关蛋白发生甲基化,进而激活其下游靶基因的转录并促进CRPC的生长。Kim等[21]发现,在胶质瘤干细胞中,激活的EZH2可与转录因子STAT3结合进而使STAT3蛋白在第180位的赖氨酸发生甲基化,甲基化的STAT3可增强其赖氨酸磷酸化的活性,最终激活其下游基因的转录。
此外,Lee等[22]的研究结果显示,EZH2与视网膜相关孤核受体α(RAR-related orphan receptor α,RORα)结合并使RORα蛋白第38位的赖氨酸发生甲基化,从而促进泛素连接酶复合物介导的RORα泛素化并导致RORα下游靶基因的沉默。这与EZH2引起STAT3、AR及AR相关蛋白甲基化进而激活靶基因的转录作用正好相反[20-21]。
2.4 不依赖于PRC2的转录激活
EZH2还可以通过不依赖于PRC2复合物而直接与多种其他因子形成转录复合物(图2),从而激活下游靶基因的转录,该作用与EZH2的酶催化活性无关[23-25]。Lee等[23]的研究表明,在雌激素受体(estrogen receptor,ER)阴性的基底样乳腺癌细胞中,EZH2与核因子κB(nuclear factor-kappa B,NF-κB)组分RelA/RelB结合形成三元复合物,进而激活NF-κB靶基因的表达。Li等[24]的结果显示,在ER阳性的乳腺癌细胞中,EZH2与TRIM28、染色质重塑蛋白SWI/SNF结合形成复合物,激活乳腺癌干细胞维持和导致不良预后的相关基因。
此外,Yan等[26]发现,JAK3会引起EZH2中第244位丝氨酸发生磷酸化,在自然杀伤/T细胞淋巴瘤中,磷酸化的EZH2可以作为转录激活因子激活增殖相关基因,该作用独立于EZH2的酶催化活性。
3 EZH2在肿瘤中的作用
3.1 EZH2与肿瘤细胞增殖
在黑色素瘤[13]、乳腺癌[15]、肺癌[27]等多种高度增殖的肿瘤中,EZH2的表达明显高于癌旁组织或正常组织,且与肿瘤患者的预后密切相关。在DLBCL[28]、滤泡性淋巴瘤[29]等淋巴瘤中,EZH2的SET催化结构的Y641和A677等位点发生功能获得性突变,使H3K27me3的水平高于正常细胞,导致肿瘤的恶化[14]。EZH2的异常过表达或功能获得性突变可以通过H3K27me3沉默相关的肿瘤抑癌基因,从而调控多条肿瘤信号通路,包括VEGF-A/AKT信号通路[30]、mTOR信号通路[31]、TGF-β-Smad-ASCL1信号通路[32]等,进而导致肿瘤细胞的恶性增殖。目前,已有大量研究表明,敲低EZH2(siRNA或shRNA法)、敲除EZH2(CRISPR/Cas9法)及抑制EZH2可以明显抑制多种肿瘤细胞的增殖,并在小鼠移植瘤模型中抑制肿瘤的生长[5, 33-34]。
3.2 EZH2与肿瘤细胞转移
近年来,EZH2在肿瘤转移中的作用及机制引起广泛关注。与非病变组织相比,EZH2在浸润性和转移性的黑色素瘤中过表达,可导致肿瘤细胞中的H3K27me3水平明显增加[35]。且EZH2的异常表达与黑色素瘤的转移和不良预后密切相关,因此,EZH2可作为治疗黑色素瘤转移的有效靶点[13, 35]。Qu等[36]和Hirukawa等[37]的研究结果显示,敲除EZH2 可以明显抑制乳腺癌细胞的骨转移和肺转移。此外,Yomtoubian等[15]的研究结果表明,EZH2过表达的肿瘤细胞具有较强的侵袭和转移能力,而EZH2催化活性失活(由CRISPR/Cas9技术构建EZH2 SET结构域突变引起),可以明显抑制乳腺癌细胞的肺部转移。
3.3 EZH2与肿瘤细胞耐药
肿瘤细胞对化疗药物耐药是众多肿瘤复发的重要原因之一。铂类药物是治疗卵巢癌的一线药物,多数患者对铂类药物的初始治疗敏感,但近80%的卵巢癌患者在2年内会出现肿瘤复发和化疗耐药。Sun等[38]最新的研究表明,顺铂给药后可通过C-Myc/miR-137轴激活EZH2表达,而EZH2的过表达可以激活卵巢癌细胞存活,从而导致卵巢癌细胞产生顺铂耐药。Wu等[39]的研究显示,EZH2可以通过沉默雌激素受体α(ERα)辅因子GREB1导致他莫昔芬耐药。在接受他莫昔芬治疗的肿瘤样本中,EZH2和GREB1的表达水平呈负相关,可作为预测患者对内分泌治疗反应的重要指标。Bai等[40]发现,抑制或敲除EZH2可以克服前列腺癌细胞对恩杂鲁胺的耐药性。机制研究表明,在恩杂鲁胺耐药的细胞中,EZH2通过与前列腺特异性抗原启动子直接结合进而抑制前列腺特异性抗原的表达。
4 EZH2抑制剂的研究进展
4.1 通过抑制SAH水解酶而间接抑制EZH2的抑制剂
2009年,Miranda等[41]发现第一个EZH2抑制剂3-deazaneplanocin A(DZNep),该化合物是一种SAH水解酶抑制剂,可以间接抑制SAM依赖的甲基转移酶EZH2的酶催化活性。因此,DZNep能够抑制多种甲基转移酶的催化作用,对EZH2的活性抑制没有选择性[42]。
4.2 与SAM竞争性结合SET活性位点的高选择性抑制剂
SET结构催化域是EZH2发挥酶催化活性功能的重要结构域,SAM为EZH2发挥催化的甲基供体。因此,竞争性抑制SAM与SET结构域结合可有效抑制EZH2介导的催化活性[5]。2012年以来,多种该类型抑制剂被开发,其报道的结构类型比较多(表1),包括:EI1[43]、GSK126/GSK2816126[44]、GSK343[45]、GSK926[45]、EPZ005687[46]、EPZ6438/E7438(tazeme-tostat)[47]、EPZ011989[48]、ZLD1039[49]、CPI-1205[50]、PF-06821497[51]等。该类抑制剂具有较好的活性和较高的选择性,如GSK126可明显抑制野生型和Y641突变型EZH2的活性,而对EZH2同源家族蛋白EZH1的抑制作用和对其他多种甲基转移酶的抑制作用较弱,分别不及对EZH2抑制活性的1/150和1/1 000[44]。Epizyme公司开发的EPZ6438同样具有较高的EZH2抑制选择性,35倍高于EZH1,4 500倍高于其他甲基转移酶[47]。
目前已有多种该类EZH2抑制剂已经或者正在进行临床试验研究[5],包括EPZ6438/E7438(tazemetostat,Epizyme公司)[52-53]、GSK126/GSK2816126(葛兰素史克公司)[54]、CPI-1205(Constellation药业)[50]、PF-06821497(辉瑞制药)[51]和SHR2554(江苏恒瑞医药),适应症包括滤泡性淋巴瘤、非霍奇金淋巴瘤、DLBCL等血液系统恶性肿瘤和横纹肌瘤、肉瘤、神经系统肿瘤、肝癌和CRPC等实体瘤[5,6,50,52-54]。2020年初,美国FDA批准EPZ6438(tazemetostat)用于晚期或转移性上皮样肉瘤的治疗,并通过快速审批途径允许EPZ6438用于EZH2突变的DLBCL和滤泡性淋巴瘤的治疗[7,55]。
4.3 EZH1/EZH2双靶点抑制剂
EZH1作为EZH2同源家族蛋白,可参与介导PRC2依赖的H3K27me3,同时也具有组蛋白甲基转移酶活性[56]。考虑到EZH1或EZH2单个催化活性丧失就可以阻止肿瘤的发生发展,因此推测EZH1/EZH2双靶点抑制剂可能具有更好的抗肿瘤作用。2013年,Konze等[57]发现了第一个口服EZH1/EZH2双靶点抑制剂UNC1999,该化合物可以同时抑制EZH1、野生型和Y641突变型EZH2的活性,而不影响其他靶蛋白。UNC1999能降低细胞内H3K27me3的水平并选择性抑制Y641突变的DLBCL的生长[57]。2017年,Honma等[58]介绍了可口服的EZH1/EZH2双靶点抑制剂OR-S1和OR-S2。OR-S1和OR-S2在Y641N突变的DLBCL中表现出较强的体内外抗肿瘤活性,并且通过减少白血病干细胞的数量延长白血病患者的生存期[59]。此外,EZH1/EZH2双靶点抑制剂DS-3201b在多种B细胞和T细胞非霍奇金淋巴瘤的临床研究中也表现出一定的抗肿瘤活性[60]。但是,除了组蛋白甲基转移酶活性,EZH1在发育、天然免疫以及干细胞多功能性的维持中也发挥着重要的作用,导致EZH1/EZH2双靶点抑制剂存在一定的潜在毒副作用。
4.4 EZH2降解剂
鉴于EZH2还可以通过PRC2非依赖性和非酶催化活性的方式发挥作用,且多种肿瘤对EZH2酶催化活性抑制剂不敏感,因此,EZH2降解剂将会成为一种抑制EZH2的新策略。靶向蛋白水解的嵌合体(proteolysis targeting chimeras,PROTACs)和疏水性标签(hydrophobic tagging,HG)是高效的选择性降解靶蛋白的技术/策略[61-62]。
2019年,Ma等[33]使用HG技术开发出第1个EZH2选择性降解抑制剂MS1943。MS1943可以显著降低多种三阴性乳腺癌细胞中EZH2的表达,并诱导细胞产生凋亡,有效抑制肿瘤细胞的增殖。敲低/敲除EZH2基因与MS1943抑制细胞增殖的作用一致,且MS1943通过靶向降解EZH2可完全抑制体内肿瘤的生长。RNA-seq实验结果显示,EZH2降解可以引起MS1943敏感性细胞中持续发生内质网应激反应而导致未折叠蛋白响应途径持续过度活化,进而导致细胞产生凋亡。
2019年底,Hsu等[63]和Potjewyd等[64]几乎同时报道靶向EED的PROTACs分子可以有效降解PRC2复合物中的EED、EZH2和SUZ12蛋白。Hsu等研究者所在的阿斯利康公司(AstraZeneca)发现的PROTAC #1和#2化合物,通过与靶蛋白EED结合,并与E3泛素连接酶一起促进三元复合物的形成,引起PRC2复合物中EED、EZH2和SUZ12蛋白的快速降解[63]。PROTAC #1和#2化合物可以有效抑制EZH2依赖性肿瘤细胞的增殖[63]。Potjewyd等合成的UNC6852也是一种靶向EED的PROTAC分子,分别与EED和VHL结合,从而诱导PRC2组分(EED,EZH2和SUZ12)的蛋白酶体降解。UNC6852降解PRC2会抑制EZH2的甲基化转移酶活性,从而降低细胞中H3K27me3的水平。UNC6852还可以降解野生型和突变型EZH2,并表现出明显的抗增殖作用。2021年,Liu等[65]报道了直接靶向EZH2降解PRC2复合物的PROTACs分子。其中,PROTAC分子E7分别与EZH2和E3连接酶小脑蛋白(cereblon,CRBN)配体结合,通过泛素蛋白酶体途径降解PRC2的核心亚基(EZH2、EED、SUZ12和RbAp48),在EZH2催化功能和非催化功能驱动的肿瘤细胞中均展现出很好的增殖抑制作用。
此外,Wang等[66]发现,天然产物新藤黄酸(gambogenic acid,GNA)及其衍生物GNA002不仅可以与EZH2共价结合抑制其催化活性,还可以引起CHIP介导的EZH2蛋白泛素化降解,进而完全抑制EZH2的致癌功能。Zhang等[67]的最新研究表明,EZH2共价抑制剂SKLB-0335(表1)与EZH2蛋白的SAM口袋共价结合,抑制底物H3K27me3的表达水平,且在药物洗脱后表现出持续抑制作用。SKLB-0335展现出优异的EZH2选择性,对EZH1下游基因未表现出明显调控作用,有别于EZH2/EZH1双靶点抑制剂,潜在毒副作用较小,具有良好的开发应用前景。
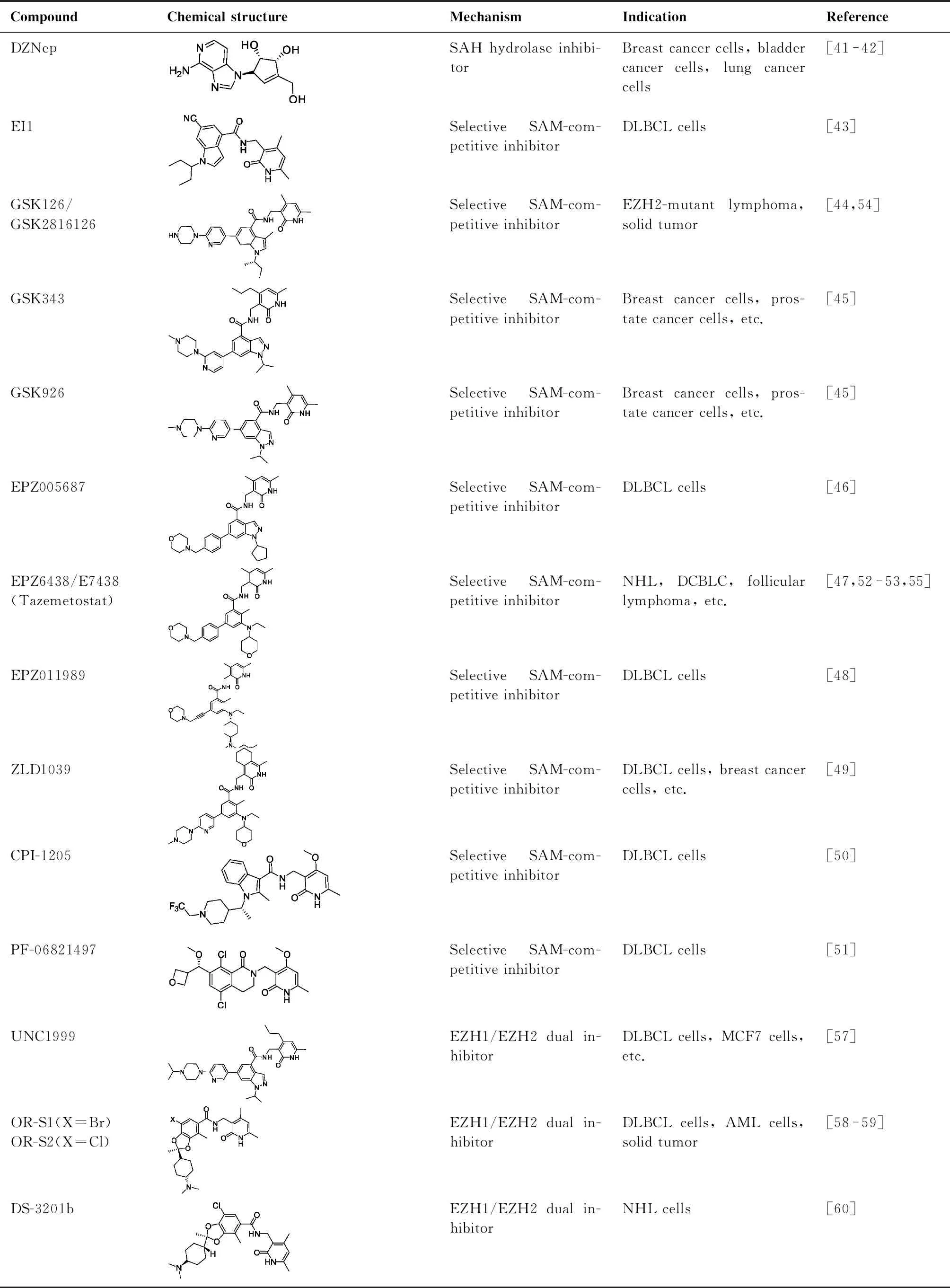
表1 EZH2抑制剂化学结构、作用机制及适应症汇总

(Table 1 continues on next page)
5 展望与讨论
EZH2作为肿瘤治疗的一种新型有效靶点,已成为众多研究的热点。EZH2可促进多种肿瘤细胞的增殖、转移和耐药。6种EZH2抑制剂相关的35项临床试验研究正在进行中,但多数仍处于临床前研究阶段或I/II期临床试验研究。目前,FDA唯一批准的EZH2抑制剂是tazemetostat (TazverikTM) ,用于晚期或转移性上皮样肉瘤的治疗。虽然EZH1/EZH2双靶点抑制剂具有更强的抗肿瘤效果,但潜在的毒副作用可能更大,已有EZH2抑制剂对EZH1的选择性有待提高,且存在给药剂量大的缺点,因此,低毒、高选择性抑制剂仍是未来EZH2抑制剂开发的主要方向。
目前,多数EZH2抑制剂主要是SAM竞争性结合SET活性位点的选择性抑制剂,考虑到EZH2还可以通过PRC2非依赖性和非酶催化活性的方式发挥作用,EZH2降解剂或同系高选择性EZH2抑制剂将成为抑制EZH2的新策略。虽然已有EZH2降解剂在开发,但其结构多样性有待丰富,且需要进一步的临床试验以评估其活性。此外,EZH2抑制剂与其他疗法联合使用已经成为肿瘤治疗的新思路,且已有数据表明EZH2抑制剂与免疫治疗或者化学治疗具有协同作用,相关研究已进入临床试验阶段。总之,EZH2抑制剂在肿瘤的临床治疗中已经取得里程碑式的成功,但仍需进一步的深入研究。
作者声明:本文全部作者对于研究和撰写的论文出现的不端行为承担相应责任;并承诺论文中涉及的原始图片、数据资料等已按照有关规定保存,可接受核查。
学术不端:本文在初审、返修及出版前均通过中国知网(CNKI)科技期刊学术不端文献检测系统的学术不端检测。
同行评议:经同行专家双盲外审,达到刊发要求。
利益冲突:所有作者均声明不存在利益冲突。
文章版权:本文出版前已与全体作者签署了论文授权书等协议。
