利用加权基因共表达网络分析方法识别帕金森病进展过程中的关键基因
2021-11-03王树文樊冬梅陈方正谢玉平吕晓彤李依璘林鸿程广东医科大学广东东莞523808
王树文,樊冬梅,陈方正,谢玉平,吕晓彤,李依璘,林鸿程 (广东医科大学,广东东莞 523808)
加权基因共表达网络分析(WGCNA)是探究基因复杂调控网络最常用到的生物信息学分析方法[1-3]。帕金森病(PD)作为一种涉及多基因调控的疾病,目前利用WGCNA 算法对PD 进行研究的文献报道不多。本研究通过构建加权基因共表达网络最终识别出PD 密切相关的关键基因模块及关键基因,以期为阐明PD 发生发展机制及治疗提供新的研究靶点分子。
1 材料和方法
1.1 PD数据集和样本信息的获取
PD相关表达谱芯片数据与样本信息下载于GEO数据库(https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE22491),芯片数据编号:GSE22491,芯片平 台:GPL6480(Agilent-014850 Whole Human Genome Microarray 4x44K G4112F),检测细胞类型:peripheral blood mononuclear cells。数据集样本信息主要包括8例正常样本和10例PD 样本信息。基于R软件中的“GEOquery”下载数据集矩阵文件和平台注释文件[4],根据注释文件对下载的矩阵文件进行基因重注释后,过滤掉注释信息缺失的探针和低表达的探针,最后利用R 包“limma”中的“normalizeBetweenArrays”函数对芯片表达谱数据进行标准化处理[5]。另外对矩阵文件中的样本信息单独整理出用于后续WGCNA 分析。为保证WGCNA 分析过程中运行的顺利性和可靠性,本研究选取了芯片表达谱中方差变化最大的前25%的基因用于下游分析。另外在正式进行WGCNA分析前,还需要通过计算样本间的相关性,绘制样本的分层聚类图,删除离群样本。
1.2 WGCNA分析
WGCNA 分析完成主要是基于R 软件中的“WGCNA”包实现[6],具体流程包括:根据表达谱计算出适宜的软阈值,在计算出两两基因之间相互强度的基础上,对其进行加权计算并构建出基于芯片表达谱的邻接矩阵,随后根据TOM 重叠计算公式将邻接矩阵转换为拓扑重叠矩阵(TOM),根据聚类树算法和1-TOM 计算公式,将表达相似基因合并在一起从而构建出多个基因集模块,同一模块内基因往往具有相似和相关的生物学功能。最后计算基因模块的特征值(ME),同时关联样本信息(正常与PD),使用“Pearson”相关系数计算模块特征值与样本性状的相关程度,将与PD相关程度最强的模块定义为PD关键基因模块,随后对该模块内基因进行深入挖掘和功能分析。
1.3 关键基因的筛选
首先计算PD 关键基因模块内MM 值和GS 值,MM 值是基因表达水平和ME 值之间的相关系数,相关性越高,说明基因和该模块的关联性就越高,GS表示每个基因与性状的相关程度,参考其他文献我们将PD 关键基因模块内基因连接度最大的前10 个基因定义为PD 关键基因,后续通过文献检索,对PD 关键基因进行进一步筛选,识别出目前在PD 研究中还未涉及到的关键基因。
1.4 基因功能(GO)和京都基因与基因组百科全书(KEGG)富集分析
利用R 软件中的“clusterprofiler”包[7]对PD 关键模块内基因进行进行GO 功能富集分析和KEGG 通路富集分析和可视化,分析结果中P<0.05 表示富集结果的差异具有统计学意义。其富集结果以气泡图形式呈现。
1.5 PD关键基因差异分析与共表达分析
利用R 软件中的“limma”包进行筛选出的10 个PD 关键基因在正常组织样本与PD 样本差异性,差异条件设置为:差异倍数绝对值≥1.2 且较正后的P<0.001。最后利用R软件中内置的“cor”函数进行10个PD关键基因的相关性分析,分析方法选用“Pearson”。
1.6 PD相关基因的筛选
基于Genecards 数据库(https://www.genecards.org/)筛选目前比较明确的PD 相关基因,从而对本研究筛选出的10个PD关键基因进行初步验证。
2 结果
2.1 芯片表达谱预处理及加权基因共表达网络的构建
整理GSE22491 芯片表达谱,过滤其中探针信息注释不全和重复的基因,最终获得16 576 个基因,其中包括16 327个编码基因和249个长非编码RNA,选取方差变化最大的前4 082 个基因进行后续的WGCNA 分析。通过计算样本间的相关性,绘制样本的分层聚类图,将聚类高度设置为90(红线标记),见图1。删除异常样本GSM558687,剩余17 例样本信息及相应的4 082 个基因芯片表达谱数据合并用于后续WGCNA构建模块,见图2。

图1 样本聚类并筛选异常样本(红线表示高度=90)

图2 样本聚类与对应的样本信息
2.2 加权基因共表达网络的构建
基于文献中WGCNA 分析流程,借助R 软件中“WGCNA”包实现本研究所需的加权基因共表达网络的构建。在WGCNA分析过程中,软阈值的选取对于构建符合生物学意义的无尺度网络具有决定性的作用,本研究基于“WGCNA”包中的“pickSoftThreshold”函数进行了软阈值的筛选,见图3。当选软阈值即β=7 时,可以保证本研究构建出的网络既达到无尺度标准,也使网络的平均连接程度不至于较小而缺乏足够的信息。随后计算基因间的相关性矩阵和邻接矩阵,根据TOM 重叠计算公式将邻接矩阵转换为TOM 重叠矩阵,根据1-TOM 公式构建基因间分层聚类树,同时使用动态剪切树的算法把基因分成15个模块,根据模块相似度大于0.75标准合并相似模块(图4),最终基于表达谱数据可以识别出15个基因模块,包括:Black 模块(190 个基因)、Blue 模块(576 个基因)、Brown模块(430个基因)、cyan模块(130个基因)、Green 模块(257 个基因)、Greenyellow 模块(149 个基因)、Grey 模块(109 个)、Magenta 模块(158 基因)、pink模块(162个基因)、purple模块(150个基因)、Red模块(230 个基因)、samon 模块(140 个基因)、tan 模块(149个基因)、Turquoise 模块(937 个基因)和Yellow 模块(315个基因)。鉴于Grey模块内包含的基因是指不能归类于其他任何模块的基因,后续研究中一般将Grey模块进行舍弃。

图3 构建WGCNA分析过程中软阈值筛选
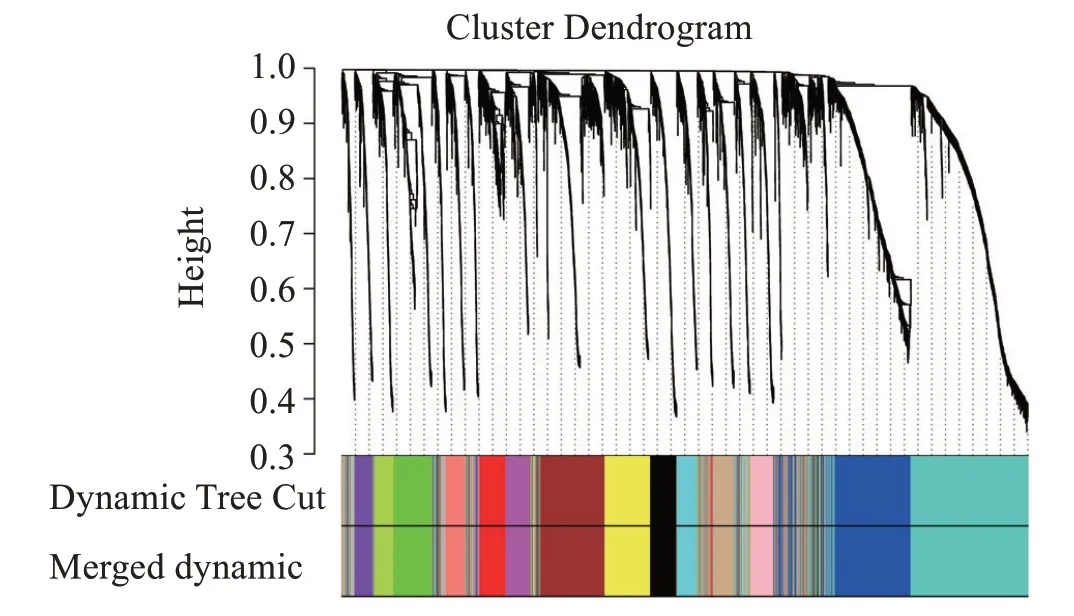
图4 基因共表达网络分层聚类树与共表达模块
2.3 与PD 样本表型的相关基因模块识别及该模块内基因功能和通路富集分析
基于Pearson相关性分析方法,通过计算各个基因模块与样本表型之间的关系,绘制出了基因模块与样本表型热图,可以看出模块Turquoise(ME-turquoise)与PD 样本显著相关(r=0.97,P=3e-10),见图5、6。随后对模块Turquoise 内937 个基因进行功能注释和通路富集分析,结果显示GO 功能注释BP 条目显著富集在氯离子跨膜转运过程、氧运输、阴离子转运和中性粒细胞生长及代谢等生物学过程,见图7。KEGG 通路富集分析显示937 个基因显著富集在神经活动配体与受体相关作用和细胞因子-细胞因子受体相互作用等通路途径。
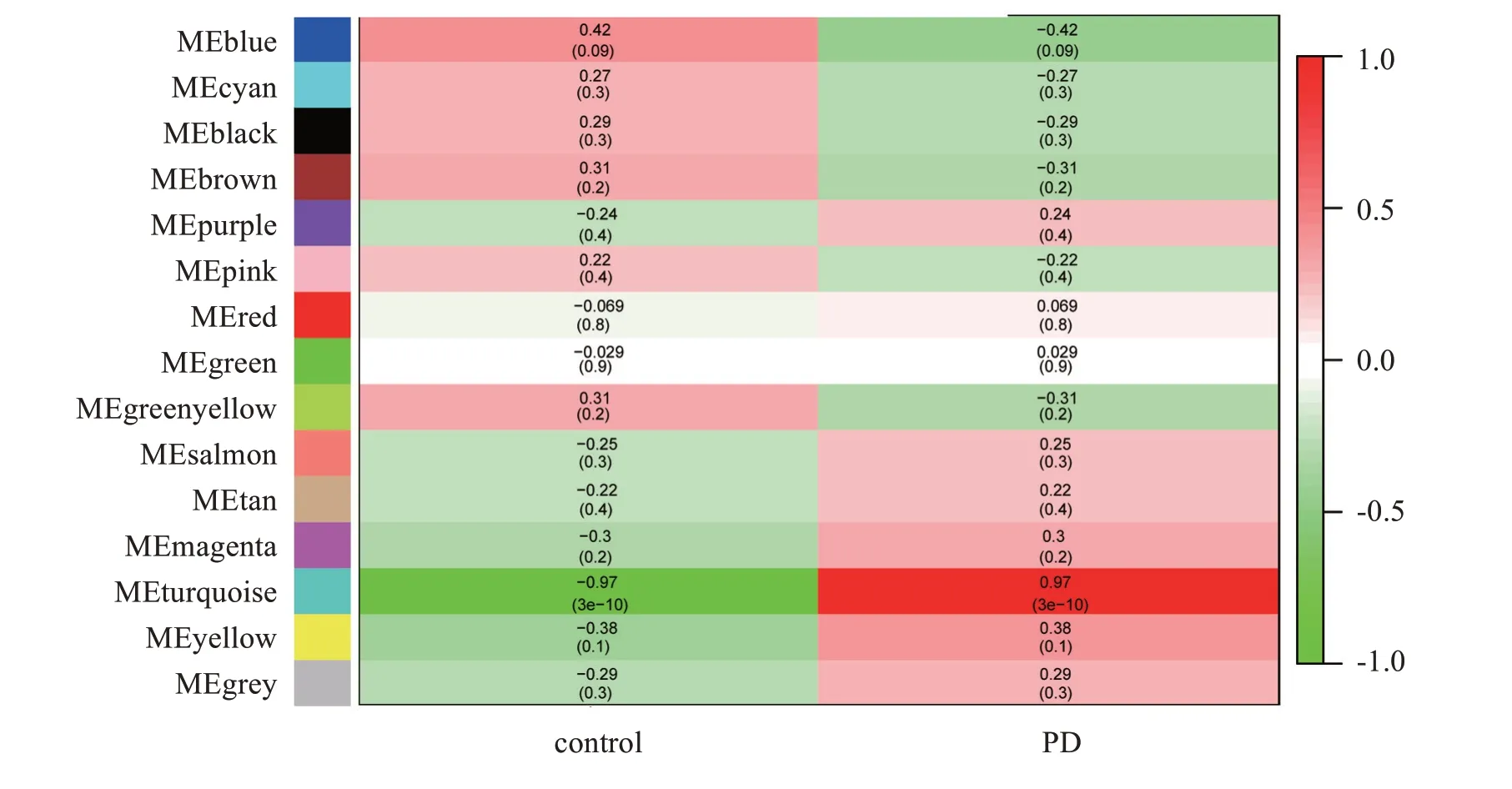
图5 WGCNA构建模块(ME值)与样本信息相关热图

图6 turquoise 模块内基因MM 与GS 散点图(红线表示GS=0.9,MM=0.9)

图7 turquoise模块内基因GO生物学过程和KEGG通路分析
2.4 PD表型相关关键基因识别
基于上述相关性分析识别出与PD显著相关基因模块,参考其他文献我们将PD 表型最相关的Turquoise 基因模块内基因基因连接度最大的前10 个基因定义为PD 关键基因,包括IFITM5、MYPOP、SIK1、NPAS3、SMARCC2、FBXO17、SLC34A3、AMN、TNRC18 和FBRSL1,其中基因IFITM5 和FBRSL1 在Genecards 数据库中已经证实参与PD 进程,而剩余8个基因与PD相关性的研究尚未见文献报道。
2.5 PD表型相关关键基因差异分析及相关性分析
利用R 软件中的“limma”包对上述筛选出的10个PD 关键基因在正常组织样本与PD 样本差异性进行分析,结果显示,与正常组织相比,筛选出的10 个PD 关键基因在PD 样本显著上升(差异倍数大于1.2且矫正后P<0.001)。相关性分析显示10 个PD 关键基因具有强的相关程度(r>0.9),见图8。
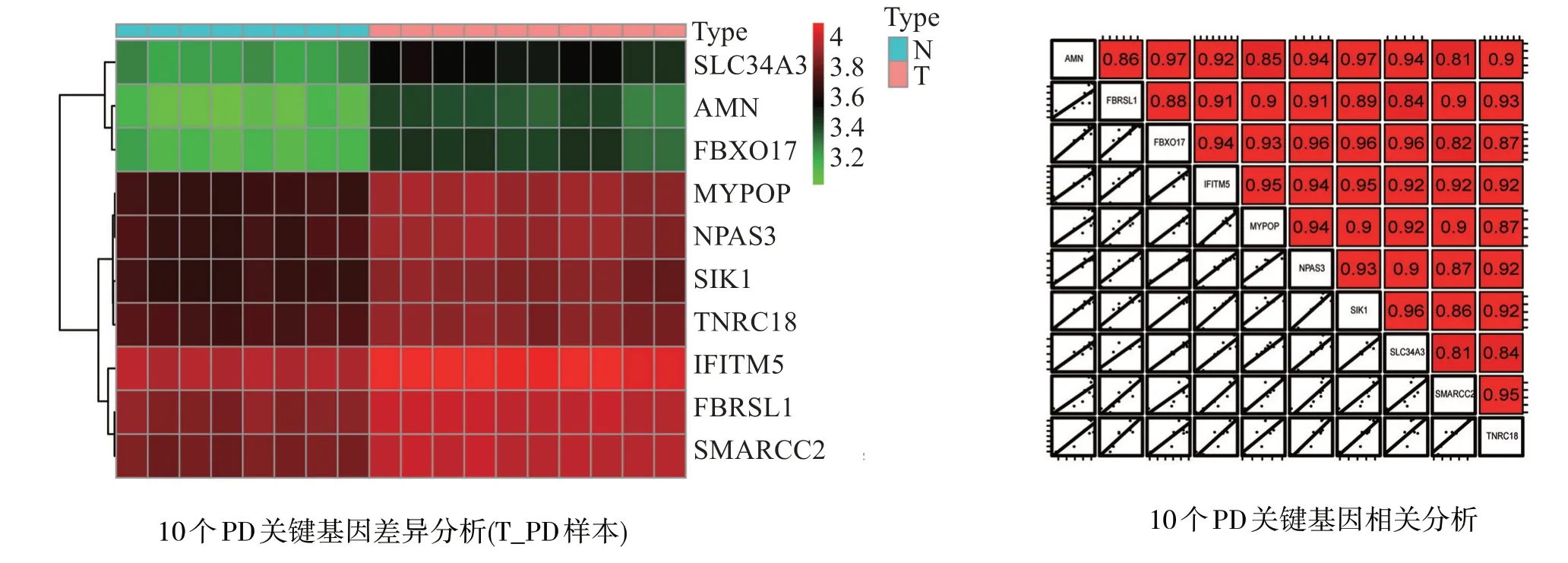
图8 10个PD相关关键基因差异分析和共表达分析
3 讨论
PD 是1 种多发于中老年人、以运动障碍为主要表现的神经系统变性疾病[8]。随着社会老龄化的日益加剧,PD 已成为我国仅次于阿尔茨海默病的神经系统性疾病[9]。目前,PD的发病机制仍存在诸多未知且临床上缺乏有效的治疗药物。因此,开展PD 分子机制的相关研究对改善目前PD现状具有积极意义。
PD 最主要的病理改变比较明确,首先由于中脑黑质多巴胺(dopamine,DA)能神经元的变性死亡,继而引起纹状体DA 含量显著性减少而导致患者致病[10]。目前关于PD 临床特征和发病机制并未明了,积极探索PD发生发展机制对于人们掌握其主要临床特征,筛选有效的药物治疗靶点具有重要意义。
多基因参与了疾病的发生发展过程,PD 也不例外。近年来出现的高通量测序技术为人们研究多基因在疾病进程中的作用提供了必要条件,相较于传统的单个分子的生物学研究,基于高通量的生物信息学研究从系统生物学的角度实现了对疾病相关多个分子靶点及其功能的探究。WGCNA 分析算法是目前进行基因共表达网络研究最常用的生物信息学分析方法,广泛用于癌症研究领域,其稳定性和有效性在多项研究中得到证实,该算法的优势是在计算两两基因相互关系的基础上引入加权值,从而构建出了更符合生物学意义的基因共表达网络。在本研究中,我们首先将WGCNA 算法用于PD 转录组基因分析,试图利用先进的生物信息学分析方法,通过对PD 高通量芯片数据的挖掘,为人们深入探究PD 的发病机制提供新的研究思路和必要的候选靶点分子。
通过构建加权基因共表达网络,我们识别出Turquoise 模块内基因与PD 具有非常强的相关性,通过对模块内基因进行功能注释和通路富集分析,结果显示该模块内基因显著富集在氯离子跨膜转运过程、氧运输、阴离子转运和中性粒细胞生长及代谢相关生物学过程和神经活动配体与受体相关作用与细胞因子-细胞因子受体相互作用等通路途径,其中已有文献报道[11-12],功能和通路中相关分子参与了PD 进展的过程,本研究结论与前述文献报道较一致。我们随后对Turquoise 模块内基因进行挖掘,最终识别出10 个基因,包括IFITM5、MYPOP、SIK1、NPAS3、SMARCC2、FBXO17、SLC34A3、AMN、TNRC18 和FBRSL1,在Turquoise模块内具有MM值和GS均大于0.9,且网络连接度较高的特征。我们将上述10 个基因定义为PD 相关的关键基因。与正常样本相比,10 个基因在PD 组织中的表达均显著升高。进一步文献检索发现,此10 个基因目前在与PD 相关的研究中均较少出现。利用genecards 数据库检索此10 个基因与PD 的相关性得知,IFITM5 基因和FBRSL1 在genecards 数据库与PD均具有一定的相关性,其中IFITM5基因即干扰素诱导跨膜蛋白5,作为干扰素诱导跨膜蛋白家族成员之一,在成骨细胞矿化过程中的具有重要作用。鉴于有学者进行骨髓基质细胞向神经细胞分化及对PD 的治疗研究[13],我们推测IFITM5 可能通过影响骨髓基质细胞向成骨细胞和神经细胞的转化过程环节参与了PD 的发生、发展过程,尽管在genecards数据库中检索到FBRSL1 基因属于PD 相关基因,但其具体机制并不明确。考虑到WGCNA 构建同一模块内基因具有相似性的思路,我们对此10 个基因进行了相关性分析,结果显示,9 个基因间均与IFITM5具有非常强的相关性,其参与PD 进程的具体机制值得进一步探索。
综上所述,本研究利用WGCNA分析方法识别出了与PD具有强相关性的基因模块。通过对该模块内基因进行挖掘,识别出10 个与PD 强相关性的关键基因,但目前相关研究仍非常缺乏。因此,本研究的完成将会为进行PD 致病机理的相关研究及PD 药物靶点研究提供重要的候选靶点分子。
