Age and genotype dependent erythropoietin protection in COVID-19
2021-11-02KonstantinosPapadopoulosWarachayaSutheesophonSomjateManipalviratnTarChoonAw
Konstantinos I Papadopoulos, Warachaya Sutheesophon, Somjate Manipalviratn, Tar-Choon Aw
Konstantinos I Papadopoulos, Department of Research and Development, THAI StemLife,Bangkok 10310, Thailand
Warachaya Sutheesophon, Laboratory Department, THAI StemLife, Bangkok 10310, Thailand
Somjate Manipalviratn, Department of Reproductive Endocrinology, Jetanin Institute for Assisted Reproduction, Bangkok 10330, Thailand
Tar-Choon Aw, Department of Laboratory Medicine, Changi General Hospital, Singapore 529889, Singapore
Tar-Choon Aw, Department of Medicine, National University of Singapore, Singapore 119228,Singapore
Abstract Erythropoietin (EPO) is the main mediator of erythropoiesis and an important tissue protective hormone that appears to mediate an ancestral neuroprotective innate immune response mechanism at an early age. When the young brain is threatened-prematurity, neonatal hyperbilirubinemia, malaria- EPO is hypersecreted disproportionately to any concurrent anemic stimuli. Under eons of severe malarial selection pressure, neuroprotective EPO augmenting genetic determinants such as the various hemoglobinopathies, and the angiotensin converting enzyme (ACE) I/D polymorphism, have been positively selected.When malarial and other cerebral threats abate and the young child survives to adulthood, EPO subsides. Sustained high ACE and angiotensin II (Ang II) levels through the ACE D allele in adulthood may then become detrimental as witnessed by epidemiological studies. The ubiquitous renin angiotensin system(RAS) influences the α-klotho/fibroblast growth factor 23 (FGF23) circuitry, and both are interconnected with EPO. Here we propose that at a young age, EPO augmenting genetic determinants through ACE D allele elevated Ang II levels in some or HbE/beta thalassemia in others would increase EPO levels and shield against coronavirus disease 2019, akin to protection from malaria and dengue fever. Human evolution may use ACE2 as a “bait” for severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) to gain cellular entry in order to trigger an ACE/ACE2 imbalance and stimulate EPO hypersecretion using tissue RAS,uncoupled from hemoglobin levels. In subjects without EPO augmenting genetic determinants at any age, ACE2 binding and internalization upon SARS-CoV-2 entry would trigger an ACE/ACE2 imbalance, and Ang II oversecretion leading to protective EPO stimulation. In children, low nasal ACE2 Levels would beneficially augment this imbalance, especially for those without protective genetic determinants. On the other hand, in predisposed adults with the ACE D allele,ACE/ACE2 imbalance, may lead to uncontrolled RAS overactivity and an Ang II induced proinflammatory state and immune dysregulation, with interleukin 6 (IL-6), plasminogen activator inhibitor, and FGF23 elevations. IL-6 induced EPO suppression, aggravated through co-morbidities such as hypertension, diabetes,obesity, and RAS pharmacological interventions may potentially lead to acute respiratory distress syndrome, cytokine storm and/or autoimmunity. HbE/beta thalassemia carriers would enjoy protection at any age as their EPO stimulation is uncoupled from the RAS system. The timely use of rhEPO, EPO analogs,acetylsalicylic acid, bioactive lipids, or FGF23 antagonists in genetically predisposed individuals may counteract those detrimental effects.
Key Words: Erythropoietin; Angiotensin converting enzyme; Angiotensin II; Hemoglobinopathy; Malaria; Coronavirus disease 2019; Fibroblast growth factor 23
INTRODUCTION
The severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2), the cause of the coronavirus disease of 2019 pandemic (COVID-19) has to date (September 11, 2021)infected almost 225 million people worldwide, causing nearly 4.6 million deaths[1].The COVID-19 pandemic continues to be a global threat despite increasing vaccinations[1]. We and others have recently proposed that the thalassemias and especially HbE, might confer resistance to and/or protection from SARS-CoV2 infection and severity[2,3]. Supporting this hypothesis, Litteraet al[4] from Sardinia found none of their seriously ill COVID-19 patients were carriers of beta-thalassemia while a recent metanalysis reported a pooled incidence rate of COVID-19 in patients with beta thalassemia at 1.34 per 100000 personday, which is less than half of that observed in the general population (2.89)[5]. We hypothesized that host immune system modulations engendered by malarial selection pressureviathalassemia/HbE mutations might confer this protection akin to an antimalarial effect[2]. Another genetic variant significantly associated with mild malariavssevere malaria is the D allele of angiotensin converting enzyme (ACE) I/D polymorphism, that codes for higher ACE levels and subsequently increased angiotensin II (Ang II) productionvsthe I allele[6-8]. We attempted, therefore, to trace a common denominator to explain the emergence of those two genetic determinants forced by malarial evolutionary pressure. We posit here that the evolutionary selection of thalassemias and the ACE D allele as adaptive alleles for pathogen resistance is neither coincidental nor surprising.Both genetic determinants appear to elicit and sustain a phylogenetically preserved ancestral neuroprotective innate immune response mechanism against tissue injury or pathogen invasion mediated eitherviasystemic or/and local increases in erythropoietin (EPO) production[9].
In the present review, we will attempt to explain how (1) Elevated EPO can account for COVID-19 protection in the young; (2) EPO augmenting genetic determinants can predispose for severe COVID-19 complications in adults, and (3) Endogenous and/or pharmacological EPO modulation may offer innovative approaches to treat and/or mitigate SARS-CoV-2 disease severity.
EPO’S TISSUE PROTECTIVE ACTIONS
EPO is an evolutionary conserved hormone, well known for almost a century as the main mediator of erythropoiesis but its widespread effects throughout the body might transcend its primary role[9]. EPO’s principal physiologic stimulus for secretion is tissue hypoxia which upon detection by renal interstitial cells is subsequently secreted[9]. Apart from its two main sites of secretion, the kidney and liver, EPO is locally produced and released in a paracrine or autocrine fashion by cells of various tissues including the heart, lungs, testes, ovaries, enterocytes, breast gland and human milk,spleen, bone marrow macrophages, placenta, retina, astrocytes, and neurons[10,11].EPO’s erythropoietic effects are mediatedviabinding to an EPO receptor (EPOR)homodimer (EPOR)2on erythroid precursors[9]. Evidence supports the renin angiotensin system (RAS) systemviaAng II and the EPO-fibroblast growth factor 23(FGF23) signaling pathway as additional regulatory pathways, possibly involved in EPO’s non-hematological functions[12,13]. EPO’s two distinctive activities(erythropoiesis and tissue protection) appear to reside in different EPO domains and bind to two distinct receptors[14].
When pathogen invasion, tissue trauma or insult occurs, a defensive strategic ensemble is summoned, spearheaded by chemokines and inflammatory cytokines, to attract armies of immune cells that fend off, isolate, kill and remove pathogens and dead cells. This process needs to be controlled and must not be allowed to propagate.Thus, a tissue protective mechanism is required and seems to be provided by the presence of EPOviaits binding to the tissue-protective receptor (TPR), a heteromeric complex between the EPOR and the β common receptor[9,14]. The TPR is typically not highly expressed but compartmentalized intracellularly and is up-regulated and exposed when insult, trauma, hypoxia, and inflammation invoke subsequent tissue protection[9]. It also has a much lower EPO affinity and needs as high as fivefold systemic EPO levels to be activated[9]. EPO’s tissue protective, tissue regenerative,angiogenetic, anti-inflammatory, and anti-apoptotic effects have been documentedviaexogenous EPO administration in both vertebrates and invertebrates and in a variety of disease models[11,15,16] and correlates to the expression of the EPOR in those nonhematopoietic tissues[11]. EPOviaEPOR expressed on various immune cells, can directly affect the way immune cells exert their immunoregulatory effects, and shift the function of the immune system towards suppression, swing the inflammatory response to immune tolerance, protect injured tissues from apoptosis, and promote wound healing[17]. EPO’s immunoregulatory effects have been demonstrated in experimental autoimmune encephalomyelitis[18] and in Th17 cell-associated immunemediated kidney diseasesviaEPO binding to T cell-expressed EPOR inhibiting Th17 cell induction[19]. Furthermore, EPO’s beneficial pleiotropic effects on alveolarcapillary barrier integrity in acute lung injury/acute respiratory distress syndrome(ARDS) have been proposed to be potentially mediated through EPO’s anti-inflammatory, anti-apoptotic, anti-oxidative, pro-angiogenic and cytoprotective actions[20,21]. Finally, EPO stimulates bone marrow endothelial progenitor cell mobilization possibly contributing to pulmonary endothelial repair through fusion with resident cells, paracrine effects, or combinations of both[20,21].
YOUNG AGE AND EPO AUGMENTING GENETIC DETERMINANTS:EVOLUTIONARY LESSONS ON HOW TO “SAVE THE CHILDREN”
As TPR has a much lower EPO affinity, local tissue concentrations need to be high to activate it[9].High endogenous EPO, dissonantly elevated from what is expected by a concurrent anemic stimulus and presumably to exert its non-erythropoietic tissue protective functions, has been reported in few studies[22-25]. In all these situations, an imminent tissue insult or pathogen invasion are present while young age (< 13 years)appears to be an important and independent determinant of EPO response unrelated to the circulating hemoglobin levels (Figure 1)[22-25]. Cord blood EPO levels are strongly correlated to cord blood bilirubin in pathological neonatal hyperbilirubinemia potentially shielding the newborn brain from an imminent kernicterus[23]. In extremely premature newborns, elevated endogenous EPO levels varied with circulating levels of inflammation-related proteins possibly mediating protective and repair mechanisms[24]. As a response to pathogen invasion, younger children at all degrees of severe malarial anemia (SMA), tends to have significantly higher EPO levels than expected from their degree of anemia, a phenomenon that declines with increasing age[25]. That the maximum EPO response in SMA occurred very early and at a time when cerebral malaria is uncommon reinforces the notion of an appropriate tissue protective role for EPO[25]. In that sense, the emergence of the two specific classes of malaria protective genetic determinants (the thalassemias and the ACE D allele) is congruent with the evolutionary objective of augmenting either systemic and/or local tissue EPO concentrations to mitigate tissue injury and/or pathogen invasion. The above SMA described age-related EPO pattern has also been reported in sickle cell, and HbE/β-thalassemic children without malaria[22,25]. The numerous mutations of the globin genes in thalassemias cause various degrees of anemia that are a potent and sustained stimulus for renal EPO secretion with elevated systemic EPO levels[22,25]. The ensuing ineffective erythropoiesis in thalassemias[25] avoids polycythemia and subsequent prothrombotic complications but ensures persistent and high enough EPO levels to engage the TPR in various tissues to protect against malaria and its feared cerebral complications[26]. The ACE D allele, also significantly associated with milder forms of malaria in areas of high malarial burden, is another sophisticated genetic selection[5,27-29]. Widespread RAS presence in every human organ and the presence of the ACE D allele ensure that adequate substrate, and enzyme levels (ACE) are abundant[30,31], to provide for systemically and/or locally elevated Ang II levels[7,8] sufficient for endocrine or paracrine effects on EPO secretion stimulation[12,32]. In addition, Ang II may exert immune system modulation[33] and/or direct anti plasmodium activity[34]. The subsequently increased local tissue EPO levels would thus bypass systemic EPO prothrombotic effects while possibly also conferring the demanded tissue protection[35] and mitigation against Plasmodium invasion[12,26,32]. Significantly higher age-related ACE activities in serum are found in newborns and premature infants as well as healthy children and teenagers than adults [36]. Furthermore, lower nasal ACE2 expression in children relative to adults has been reported (Figure 1)[37].
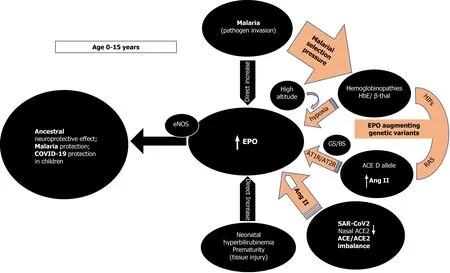
Figure 1 Age dependent erythropoietin secretion and effect of erythropoietin augmenting genetic determinants inducing ancestral neuroprotection, malaria protection, and possibly coronavirus disease 2019 protection in children. ACE: Angiotensin converting enzyme; ACE2:Angiotensin converting enzyme 2; EPO: Erythropoietin; Ang II: Angiotensin II; β-thal: Beta thalassemia; GS/BS: Gitelman syndrome/ Bartter Syndrome; HIFs: hypoxia inducible factors; SARS-COV-2: Severe acute respiratory syndrome coronavirus-2; COVID-19: Coronavirus disease 2019; RAS: Renin angiotensin system; AT1R:Ang II type 1 receptor; AT2R: Ang II type 2 receptor; eNOS: Endothelial nitric oxide synthase.
EPO IS AN ANCESTRAL NEUROPROTECTIVE MECHANISM PREVENTING LETHAL CEREBRAL INSULTS AT YOUNG AGE: IMPLICATIONS FOR COVID-19
The above findings and the presence of EPO-like signaling involved in neuroprotection in insects that lack hematopoiesis[38], reinforce the rational assumption that, in younger age groups, high EPO levels could mediate a phylogenetically preserved ancestral neuroprotective innate immune response mechanism preventing lethal cerebral damage from both non-communicable (kernicterus, prematurity)[23,24] and communicable insults (cerebral malaria) (Figure 1)[25,26]. Preliminary evidence suggests that children are indeed less likely to be symptomatic or develop severe symptoms when infected with SARS-CoV-2[39] but whether elevated EPO levels could account for the milder COVID-19 course is currently not known as EPO levels have not been reported in pediatric COVID-19 patients. It is however, known that EPO levels are significantly decreased in adult patients with critical COVID-19[40,41]. It is conceivable that evolution uses the ACE2 as a “bait” for SARS-CoV-2 to gain cellular entry in order to trigger an ACE/ACE2 imbalance[42-44] and stimulate EPO hypersecretion using RAS, uncoupled from hemoglobin levels. Low nasal ACE2 Levels present in children[37] would beneficially intensify this imbalance, especially for those without protective genetic determinants[37]. Genetically predisposed children already enjoy protective EPO levels through sustained elevated Ang II levels, through the ACE D allele in some, the ACE2 T allele leading to lower ACE2 expression in females[6,45],or HbE/beta thalassemia in others, thus protecting against coronavirus disease 2019(COVID-19), in similar ways seen in malaria and dengue fever[46] (Figure 1). EPO secretion augmenting genetic determinants alone or synergistically, might protect from or allow an asymptomatic and uncomplicated SARS-CoV2 infection leading to seropositivity and subsequent immunity[2]. In the 2ndIndian serosurvey, where only 3% of the seropositive individuals reported symptoms suggestive of COVID-19[47],the highest seropositivity rate was from the state of Odisha (formerly Orissa), where almost one quarter of the malaria burden of India is found[48]. Surreptitiously, in the same area, α-thalassemia, sickle cell and β-thalassemia alleles were found in 50.84%,13.1% and 3.4% of subjects[49], respectively while in the same geographical region, the frequency of ACE D allele was significantly higher (57.9%) in mild malaria patients as compared to those in severe malaria patients[6].
It seems intuitive to assume that endogenously increased EPO levels represent an innate “survival mode” that indeed protects the young from tissue injury and pathogen invasion. Longitudinal studies show an overall decrease in EPO levels with increasing age, but the influence of the ACE D allele/DD genotype on EPO decline is not known. Sustained and chronically elevated EPO levels in young or middle-aged non-anemic adults could herald an evolving glucose intolerance or hypertension (HT)[50,51] and later in life establish unfavorable associations with cardiovascular events[52], kidney function decline[52], fracture risk[53], and mortality[52]. Most, if not all the above conditions share associations with the ACE D allele[54] and thus, elevated EPO levels in non-anemic individuals maybe a marker for the presence of the D allele and the elevated Ang II it subsequently encodes[7,8,55,56]. The malarial protection engendered by the EPO augmenting ACE D allele[6,26-28], and the ACE2 T allele[6,45], may thus represent an evolutionary trade off and come at the expense of creating a disadvantage in older age[52] including increased risk of infection, complications, and mortality in COVID-19[45,57-59]. The association of HT with higher risk of severe or fatal COVID-19[60] and association of HT with the ACE D/ACE2 T alleles reported in several Indian populations[44,61,62] could explain the statistics observed in India during the current phase of the COVID-19 pandemic[1].
THE ACE D ALLELE / DD GENOTYPE AND EPO INTERPLAY:IMPLICATIONS FOR COVID-19
RAS and Ang II effects demonstrate impressive complexity (Figure 2)[30,31].
First, in severe acute respiratory syndrome (SARS) and COVID-19 most deaths occur due to ARDS[63]. The frequency of the ACE D allele was reported to be significantly higher in ARDS[64] but also in the hypoxemic group in Vietnamese patients with SARS related ARDS in the first SARS epidemic[65]. The association of the ACE D allele/DD genotype with increased mortality is now being increasingly reported in various ethnic groups in SARS-CoV-2 as well[59,66]. This association might reflect the effects the ACE D allele exertsviaAng II on interleukin 6 (IL-6) and plasminogen activator inhibitor-1 (PAI-1) levels (Figure 2)[67,68]. Both IL-6 and PAI-1 Levels correlate with Ang II and are the highest in individuals with the ACE DD genotype[67-70]. IL-6 can inhibit EPO secretion in the kidney[71], is a prognosticator of COVID-19 disease severity, progress to severe disease and mortality[72,73]. Similarly,elevated PAI-1 is an independent risk factor for poor ARDS outcomes in COVID-19[74]and IL-6 induced significantly elevated PAI-1 Levels in critically ill COVID-19 patients[74,75]. This suggests that the ACE gene I/D polymorphism may play important roles in SARS-CoV-2 infection disease progression into ARDS, and dysregulated immune response[59].
Congruent to its primary evolutionary (neuroprotective) objective of enhanced EPO secretion when threatened by pathogen invasion, ACE D allele/DD-genotype elevated levels of Ang II, reduce ACE2 tissue expression and activity by stimulation of lysosomal degradation through an Ang II type 1 receptor (AT1R) dependent mechanism and thus, might mitigate entry of pathogens using the ACE2 receptor[76,77]. The ACE2 malaria protective T allele could further reduce ACE2 expression and similarly mitigate pathogen entry[45]. ACE2 is ubiquitous and also present in type I and type II alveolar epithelial cells[78,79]. Loss of ACE2 expression with increasing age, in males, and type 2 diabetes (DM)[80], is known to precipitate severe acute lung failure[81]. Binding and internalization of ACE2 by SARS-CoV-1/2 involves the same AT1R dependent mechanism as Ang II[44], in reducing ACE2 cell surface expression[42,43]. A vicious circle of ACE/Ang II/ACE2 imbalance and persistently increased Ang II levels through continual RAS over-activation might lead to lung shut-down, in similar mechanistic ways as described in human H7N9[82] and H5N1[83].Additionally, an aberrant T-cell-mediated immune response and cytokine storm could be further mediated by the excessively elevated and unopposed Ang II levels[63,84].Clonally expanded tissue-resident memory-like Th17 cells have been reported in the bronchoalveolar lavage fluid from patients with severe COVID-19[85]. Th17 cells are under the influence of Ang II signaling[86] and their cell numbers were associated with disease severity and lung damage. Th17 cells demonstrate a potentially pathogenic profile of cytokine expression that may lead to immune-mediated inflammatory diseases[57,85,86]. Both EPO binding to T cell-expressed EPOR as well as AT1R block have been shown to inhibit Th17 cell induction[19,86].
Moreover, Ang II from a functional T-cell RAS plays a pivotal role in T-cell activation towards pro-inflammatory effects, proliferation, chemotaxis, cytokine production, and regulation of memory CD8+ T cell development[33,86]. All these Ang II effects could explain the adverse ACE D allele autoimmunity associations across several ethnicities and autoimmune conditions such as multiple sclerosis (MS)[86],systemic lupus erythematosus (SLE)[87,88], rheumatoid arthritis[89] and vitiligo along with higher IL-6 Levels[89-92]. In addition, Ang II induced pyroptosis, an inflammasome initiated lytic form of programmed cell death further contributes to the COVID-19 cytokine storm[93]. In COVID-19 and under the influence of the ACE D allele and the excessively increased Ang II levels[84], caspase-1 mediated pyroptotic inflammatory cell necrosis could lead to autoantigen exposure and stimulate multiple autoantibody production[94], thus leading to the development of a myriad of autoimmune conditions such as MS, SLE, antiphospholipid antibodies and syndrome,autoimmune hemolytic anemia, and thrombocytopenia, Guillain-Barré syndrome,vasculitis as well as a Kawasaki like syndrome with autoantibodies to ACE2 in children[95]. This pattern that is analogous to our findings in sarcoidosis where ACE D allele induced serum ACE increase and subsequent Ang II elevation can steer the immune system towards a protracted course with aberrant gastrointestinal immune reactivity and endocrine autoimmunity including polyglandular autoimmune syndromes[96-98]. Moreover, it has been reported that in acute sarcoidosis presenting with erythema nodosum and usually a benign and self-restricting course, the ACE DD genotype, significantly worsens prognosis[99]. Caspase-1 mediated pyroptosis and autoantigen exposure could lead to AT1R autoantibodies[94], shown to correlate significantly with IL-6[100], that can further mediate persistent proinflammatory Ang II effects by agonistic stimulation of AT1 receptors and increased AT1 receptor activity, even in the absence of the ACE D allele. Low-dose acetylsalicylic acid (ASA)[101] and increasing bioactive lipid (BAL) intake [arachidonic acid (20:4 n-6),eicosapentaenoic acid (20:5 n-3), and docosahexaenoic acid (22:6 n-3)] may result in the formation of increased amounts of endogenous Lipoxin A4 (LXA4) thus offering novel treatment options in the prevention and management of COVID-19 (Figure 2)[102].Drug design research using LXA4 as a lead compound might result to innovative treatment modalities in autoimmune diseases[94].
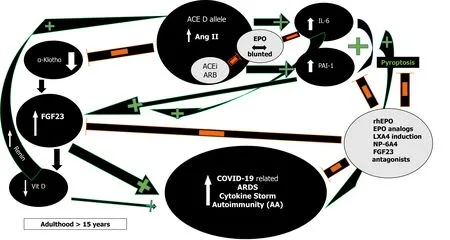
Figure 2 Proinflammatory effects of angiotensin converting enzyme D allele induced Angiotensin II via plasminogen activator inhibitor-1 and interleukin 6 induction and their effects on the α-Klotho/fibroblast growth factor 23 axis; inhibitory action of recombinant human erythropoietin/erythropoietin analogs/Lipoxin A4/fibroblast growth factor 23 antagonists. Orange minus sign denotes inhibition. Green plus sign denotes stimulation. ACE: Angiotensin converting enzyme; FGF23: Fibroblast growth factor 23; PAI-1: Plasminogen activator inhibitor-1; IL-6: Interleukin 6; rhEPO:Recombinant human erythropoietin; Vit D: Vitamin D; ARDS: Acute respiratory distress syndrome; AA: Autoantibodies; LXA4: Lipoxin A4; ACEi: Angiotensin converting enzyme inhibitors; ARB: Angiotensin receptor blockers; NP-6A4: AT2R peptide agonist.
Second, RAS influence on EPO levels likely represents an amalgam of complex,intercalated and interrelated set of signals involving multiple molecular mechanisms[12,32,103-106]. Endogenously elevated EPO levels due to hypoxia in high altitude[107,108] or in human genetic models seem protective[109] while low EPO levels are associated with dismal COVID-19 prognosis (Figure 1)[41]. Epidemiological studies suggest that physiological adaptation in a hypoxic environment at high altitude may protect persons from the severe impact of acute infection caused by SARS-CoV-2[107,108]. Reductions in cumulative incidence and mortality rates of COVID-19 with increasing altitude have been reported[107,108]. Possible explanations are related to reduced virulence and decreased SARS-CoV-2 pathogenicity at high altitude[107]along with physiological acclimatization to chronic hypoxiaviaincreased EPO and genetically adapted high altitude native populations with lower ACE DD genotype frequency[108,110]. Recently, patients with fatal COVID-19 at 4150 meters above sea level displayed 2.5 times lower EPO levels compared to survivors but Ang II levels were not measured in that study[41].
Furthermore, studies in patients with inherited genetic defects in specific kidney transporters and ion channels such as Gitelman’s and Bartter’s Syndromes (GS/BS)showed a statistically significant absence of COVID-19 infection and COVID-19 symptoms (Figure 1)[109]. In GS/BS patients, the above-mentioned genetic defects result in defective salt reabsorption in the thick ascending limb of loop of Henle[109].The resulting salt wasting, hypokalemia, and metabolic alkalosis with relatively low levels of serum chloride induce chronic RAS activation with elevated Ang II levels but due to AT1R signaling defects a hypertensive phenotype is not seen[111]. Instead,endogenously increased levels of aberrantly glycosylated ACE2[112] and Ang 1-7 counteract Ang II effects[109,112]. Intriguingly, GS/BS patients also demonstrate Ang II receptor type 2 dependent increase in EPO levels[103] and lack of Ang II induced increase of the PAI-1 gene and protein expression compared to healthy adults[113],both phenomena being possibly protective against COVID-19 at any age.
In critical and deceased COVID-19 patients, EPO levels have recently been reported to be significantly lower and not in accordance with the similarly low hemoglobin levels[40,41]. Moreover, elevated Ang II levels, strongly associated with viral load and lung injury have been reported in another study[84], and in avian influenza A virus H5N1 infected mice and H7N9 infected patients[82,83]. To date, no study has been reported in COVID-19 patients that has investigated the simultaneous measurement of Ang II and EPO and/or correlations to their ACE I/D polymorphism.
Renin and Ang II increase and RAS inhibitors inhibit EPO secretion in healthy volunteers[106]. Severe COVID-19 is also frequently associated with HT, DM, obesity,and metabolic syndrome[114], all resulting in RAS activation through various mechanisms[106]. Nevertheless, the expected Ang II induced EPO rise does not occur in critically ill COVID-19 patients even though the RAS augmenting ACE D allele may be overrepresented in both COVID-19 and associated risk diseases[58,59,61].Marathiaset al[106] recently elegantly reviewed RAS and Ang II influence on EPO secretion. Glucose and sodium reabsorption, hyperinsulinemia, the G-protein-coupled receptor 91, all induce RAS activation. The increased Ang II is expected to enhance EPO secretion through tubulointerstitial ischemia, direct upregulation of EPO transcription factors and bone marrow stimulation along with enabling erythropoiesis supportive iron metabolism[106]. On the other hand, glucose toxicity in the renal parenchyma in concurrent DM, obesity, and metabolic syndrome, induce damage on the renal EPO-producing cells and lower EPO secretion. Additionally, HT with widespread use of RAS inhibitors, diabetic hyporeninemic hypoaldosteronism,autonomic neuropathy, obesity or DM induced hypogonadism with low testosterone,chronic and acute inflammation through Ang II induced IL-6 increase[72], all inhibit renal EPO secretion (Figure 2)[71,106]. Finally, blunted EPO response has been documented in critically ill patients while a recent meta-analysis suggests that EPO therapy may decrease mortality[115].
Moreover, elevated Ang II reduces renal α-Klotho expression, interfering with FGF23 signaling and resulting in elevated FGF23 Levels (Figure 2)[116]. FGF23 will inhibit 1α-hydroxylase, leading to the lowering of 1,25-dihydroxyvitamin D3 production and cause or aggravate an incipient vitamin D deficiency, implicated in numerous adverse outcomes including morbidity and mortality in COVID-19[116,117]. All the ACE D allele associations as in HT, type 2 DM, kidney disease, and possibly mortality in COVID-19 could be explained by Ang II induced FGF23 elevations[84,116]. FGF23 serves as a proinflammatory paracrine factor, secreted mainly by M1 proinflammatory macrophages[118]. Powerful and dose-dependent associations have been demonstrated between elevated FGF23 Levels and higher risks for chronic kidney disease, left ventricular hypertrophy and congestive heart failure,autosomal dominant hypophosphatemic rickets, osteomalacia, vitamin D deficiency,fibrous dysplasia, aging, and mortality[119]. Unifying these mechanisms is the finding that both IL-6 and PAI-1 are significant regulators of FGF23 homeostasis[119-121].Dexamethasone abolished IL-6 induced FGF23 increase[119,120] while PAI-1 inhibition substantially decreased FGF23 levels (Figure 2)[121]. rhEPO administration significantly decrease PAI-1 levels in multi-trauma patients[122] and led to the miraculous recovery of a critically ill elderly COVID-19 patient[123]. EPO’s inhibitory effect on PAI-1 and subsequently FGF23 may well have contributed to the patient’s recovery and further studies are planned to investigate the potentially favorable rhEPO effect in severe COVID-19[124-126]. Human data show that both endogenous and exogenous EPO influence FGF23 levelsviaalterations of the ratio of active to inactive FGF23 in favor of its inactive form, thus attenuating effects of bioactive intact FGF23 levels and explain EPO’s protective effects[118,127]. At present, no study has been reported that investigated FGF23 levels in COVID-19.
THERAPEUTIC CONSIDERATIONS
Currently, therapeutic approaches are symptomatic and include empirical immunosuppressive and anti-inflammatory tactics (dexamethasone)[128], interferons[129], targeting of individual cytokines (IL-6: Tocilizumab/statins/heparin; PAI-1:Statins, and numerous target substances in development)[75,130-132] and correction of isolated laboratory abnormalities (e.g., sodium disturbances)[133]. Prolonged use of these interventions may lead to serious adverse effects and reduction of host defenses with resurgence of opportunistic infections.
An Occam’s razor therapeutic strategy guided by mendelian, and mechanistic evidence might be pursued. ACE I/D polymorphism genetic testing could be predictive and guide patient triage and treatment decision making as individuals with the DD genotype are predisposed to a more severe COVID-19 disease course[59].Research evidence supports the notion that endogenously[109,112] and exogenously increased EPO levels[123] could break the vicious circle of persistent ACE D allele augmented Ang II stimulation on PAI-1, IL-6 and FGF23 by both synergistic and individual inhibition[21,122,123,127,134]. Whenever the administration of rhEPO is not possible due to contraindications or heightened prothrombotic risk, EPO derivatives can coax EPO’s tissue-protective activityviaits TPR for therapeutic use without the risks attributed to EPO’s hematological actions[10,14,134]. Furthermore, EPO mediates reduction of auto-and alloantibody formation and used together with LXA4 inducing BALs and/or ASA could prevent recently reported AT1-AA induced collateral damage and autoimmune pathology[94,101,102,135,136]. Moreover, in hematologic patients, rhEPO treatment is associated with an enhanced antibody response to the influenza vaccine, similar to that of healthy subjects and it is conceivable that this effect could also be replicated in COVID-19 vaccinations, especially in immunocompromised patients[137]. Additional treatment modalities could employ a combination of autologous peripheral blood or umbilical cord-derived mesenchymal stromal cells and rhEPO/EPO derivatives that induce notable clinical improvement shortly after initiating treatment in a critically ill patient with severe ARDS[138,139].
Recently, NP-6A4, a novel AT2R peptide agonist with an FDA orphan drug designation for pediatric cardiomyopathy, increased expression of AT2R and cardioprotective EPO in a pre-clinical model with severe obesity and pre-diabetes (ZO rat), along with suppression of nineteen inflammatory cytokines including IL-6 without increasing expression levels of ACE2[140]. NP-6A4 appears as an ideal adjuvant drug candidate for EPO mediated tissue protection and mitigation of cytokine storm[140]. Finally, elucidating FGF23 Levels in COVID-19 could help prognosticate, prevent, and help treat potential future complications. The use of FGF23 antagonists such as the FGF23 antibody burosumab, could be employed to lower FGF23 Levels in FGF23-mediated disorders[141], including COVID-19. To date and to the authors’ knowledge, such clinical trials do not exist.
CONCLUSION
Age dependent EPO secretion[22-25] and the contribution of EPO augmenting genetic determinants in children and adults as a disease modifier in malaria is established[6,25-28]. In the present work, we posit that this EPO effect extends to and explains COVID-19 protection in children[39] and can provide new pathophysiological insights and therapeutic avenues in adults (Figure 1). Elevated protective EPO mRNA levels were recently reported being 2.6 times higher in nasopharyngeal swab samples of adult SARS-CoV-2 patients that were asymptomatic or showing mild COVID-19 clinical symptoms, as compared to a control group[142]. EPO induces endothelial nitric oxide (NO) synthase and increases NO production in endothelial cells[14].Increased NO bioavailability is shown to inhibit fusion of the SARS-CoV spike protein to ACE2 and early production of viral RNA [143], potentially mediating EPO protection in SARS-CoV-2 too.
The intricate balance between the components of the RAS axis (peptides and peptidases) and its interactions with the EPO and α-Klotho/FGF23 axes are incompletely understood in the context of chronic stable and acute decompensated environments. Known and unknown genetic determinants and concurrent diseases with their pharmacological interventions further complicate the view. High Ang II and low EPO levels in COVID-19, have been reported and strongly associate with viral load[84], lung injury[84], and critical disease[40,41]. Ang II, excessively augmented in the presence of the ACE D allele[7,8], leads to reduction in ACE2[44], and increases FGF23, PAI-1, and IL-6 levels[67-70,116], that along with increasing age, co-morbidities and concurrent pharmacological RAS interventions, all blunt EPO response[50,71,106]and potentially reduce EPO levels in critically ill COVID-19 adult patients (Figure 2)[40,41]. In adults with COVID-19, this proinflammatory constellation would promote progress to ARDS, and cytokine storm with pyroptotic inflammatory reactions,autoantigen exposure, autoantibody production and subsequent autoimmune disorders[95].
杂志排行

World Journal of Stem Cells的其它文章
- Translational products of adipose tissue-derived mesenchymal stem cells: Bench to bedside applications
- Unveiling the morphogenetic code: A new path at the intersection of physical energies and chemical signaling
- Alternative RNA splicing in stem cells and cancer stem cells:Importance of transcript-based expression analysis
- SOX transcription factors and glioma stem cells: Choosing between stemness and differentiation
- Retina stem cells, hopes and obstacles
- Considerations for the clinical use of stem cells in genitourinary regenerative medicine