SOX transcription factors and glioma stem cells: Choosing between stemness and differentiation
2021-11-02MilenaStevanovicNatasaKovacevicGrujicicMarijaMojsinMilenaMilivojevicDanijelaDrakulic
Milena Stevanovic, Natasa Kovacevic-Grujicic, Marija Mojsin, Milena Milivojevic, Danijela Drakulic
Milena Stevanovic, Natasa Kovacevic-Grujicic, Marija Mojsin, Milena Milivojevic, Danijela Drakulic,Laboratory for Human Molecular Genetics, Institute of Molecular Genetics and Genetic Engineering, University of Belgrade, Belgrade 11042, Serbia
Milena Stevanovic, Chair Biochemistry and Molecular Biology, Faculty of Biology, University of Belgrade, Belgrade 11158, Serbia
Milena Stevanovic, Department of Chemical and Biological Sciences, Serbian Academy of Sciences and Arts, Belgrade 11000, Serbia
Abstract Glioblastoma (GBM) is the most common, most aggressive and deadliest brain tumor. Recently, remarkable progress has been made towards understanding the cellular and molecular biology of gliomas. GBM tumor initiation, progression and relapse as well as resistance to treatments are associated with glioma stem cells(GSCs). GSCs exhibit a high proliferation rate and self-renewal capacity and the ability to differentiate into diverse cell types, generating a range of distinct cell types within the tumor, leading to cellular heterogeneity. GBM tumors may contain different subsets of GSCs, and some of them may adopt a quiescent state that protects them against chemotherapy and radiotherapy. GSCs enriched in recurrent gliomas acquire more aggressive and therapy-resistant properties,making them more malignant, able to rapidly spread. The impact of SOX transcription factors (TFs) on brain tumors has been extensively studied in the last decade. Almost all SOX genes are expressed in GBM, and their expression levels are associated with patient prognosis and survival. Numerous SOX TFs are involved in the maintenance of the stemness of GSCs or play a role in the initiation of GSC differentiation. The fine-tuning of SOX gene expression levels controls the balance between cell stemness and differentiation. Therefore,innovative therapies targeting SOX TFs are emerging as promising tools for combatting GBM. Combatting GBM has been a demanding and challenging goal for decades. The current therapeutic strategies have not yet provided a cure for GBM and have only resulted in a slight improvement in patient survival. Novel approaches will require the fine adjustment of multimodal therapeutic strategies that simultaneously target numerous hallmarks of cancer cells to win the battle against GBM.
Key Words: Glioblastoma; SOX transcription factors; Glioma stem cells; Stemness;Differentiation
INTRODUCTION
Cancer is among the leading causes of death worldwide, representing a substantial public health burden affecting welfare and life expectancy globally, with enormous impacts on individuals, families and health systems. The global burden of cancer continues to grow with its increasing incidence in the 21st century, mainly due to the growth and ageing of populations, adoption of unhealthy behaviors and exposure to unhealthy environments.
Cancers comprise a large group of diseases that arise from cells that escape the normal checkpoints of cell division and are capable of uncontrolled growth and proliferation. Deregulation of the precise molecular networks operating at the molecular and cellular levels that control cell proliferation, differentiation and cell death leads to the transformation of normal cells into cancer. Despite decades of intensive research,the underlying mechanisms that transform normal cells into cancer cells and enable cancer cells to spread and metastasize other sites in the body, leading to a fatal outcome, are still not completely understood.
The transformation of normal cells into cancer cells is a complex multistep process,and in recent decades, tremendous efforts have been made to understand the underlying mechanisms. In a highly influential article published more than 20 years ago, Hanahan and Weinberg[1] elucidated six essential biological capabilities of cancer cell, widely accepted as the six hallmarks of cancer. Most, if not all, cancers acquire this set of capabilities during the multistep development of human tumors. The six hallmarks of cancer (Figure 1) include cell sufficiency in growth signals, insensitivity to growth-inhibitory signals, evasion of programmed cell death, limitless replicative potential, sustained angiogenesis, and tissue invasion and metastasis[1]. Together,these hallmarks constitute an organized framework for interpreting the remarkable diversity of neoplastic diseases[1]. It has been proposed that the genomic instability underlying these hallmarks generates genetic diversity, which contributes to multiple cancer hallmark functions[1].
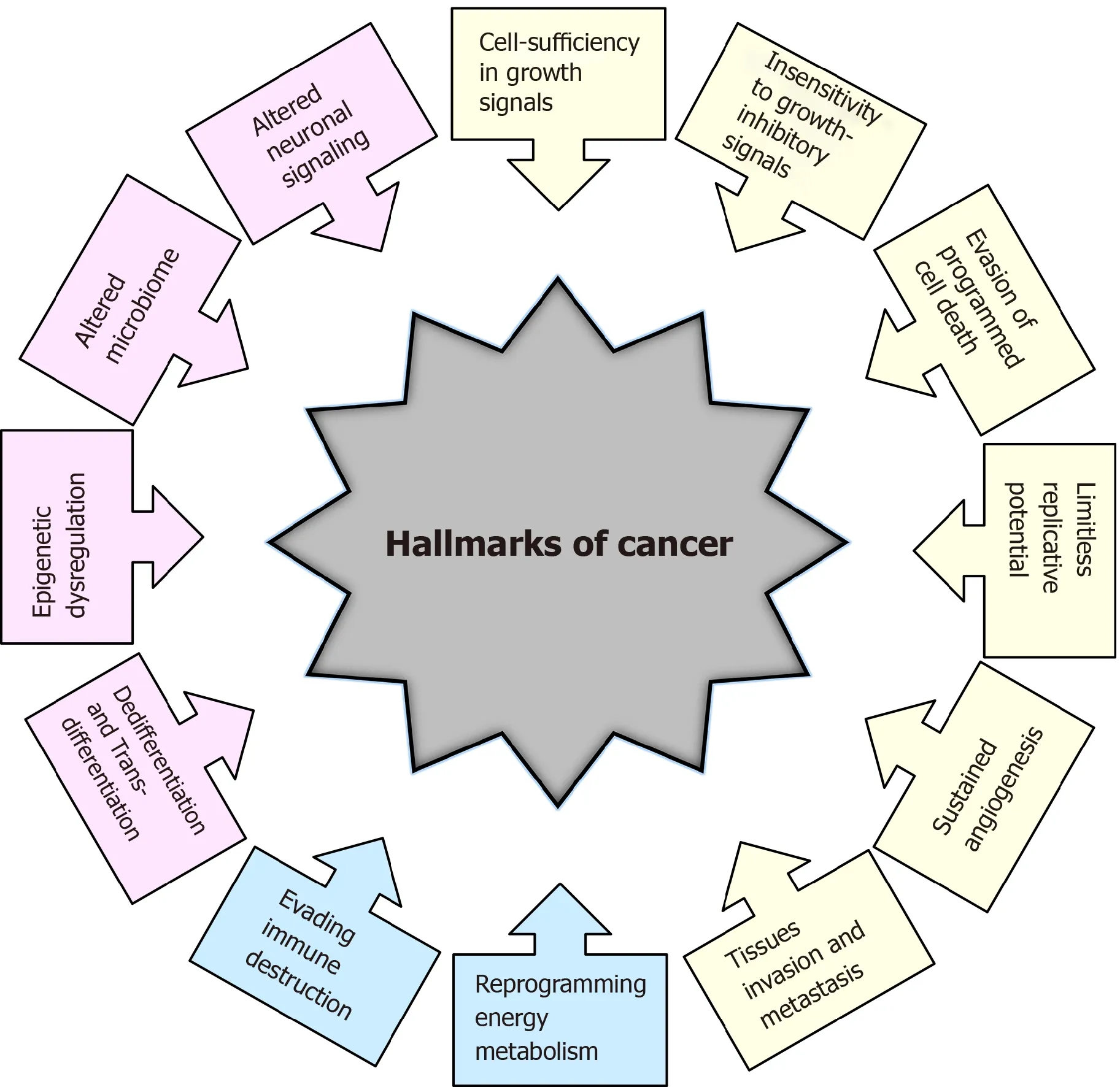
Figure 1 Historical overview of the hallmarks of cancer. Hallmarks of cancer proposed by Hanahan and Weinberg[1,2] in 2000 and 2011 are presented in yellow and blue colors, respectively. Additional hallmarks of cancer proposed by Senga and Grose[3] in 2021 are presented in pink.
In subsequent years, remarkable progress was made towards understanding the mechanisms underlying each of the six hallmarks. New technologies and significant advances in the understanding of the cellular and molecular biology of cancer cells provided better insight into the multiple events that allow cancer cells to acquire additional functional capabilities enabling them to survive, proliferate and disseminate. Thus, two additional enabling characteristics of cancer cells-genomic instability and inflammationviainnate immune cells, have been defined[2]. Genomic instability in cancer cells generates random mutations and acquired genetic diversity,endowing cancer cells with genetic alterations that drive tumor progression[3]. Inflammation that fights infections and heals wounds under physiological conditions can assist tumor progression in premalignant and malignant lesions[4]. Research on the intersections between inflammation and cancer pathogenesis has provided overwhelming evidence of the essential effects that immune cells, largely of the innate immune system, have on tumor-promoting neoplastic progression[4-6].
In an updated article published by Hanahan and Weinberg[2] in 2011, two novel hallmarks were proposed and added to the list of core hallmarks of cancer:reprogramming energy metabolism and evading immune destruction. The first includes the major reprogramming of cellular energy metabolism that is required to support uncontrolled cell proliferation. Since the immune system serves as an important barrier to tumor formation, the second hallmark implicates the ability of cancer cells to evade attack and elimination by immune cells[2].
It has become necessary for the hallmarks of cancer to be revised and upgraded as a result of the progression of cancer research and the accumulation of knowledge over the last decade. Four novel hallmarks of cancer were recently proposed, justified and incorporated into the mainstream hallmark conceptualization. These hallmarks include dedifferentiation and transdifferentiation, epigenetic dysregulation, altered microbiome and altered neuronal signaling (Figure 1)[7].
STEM CELLS: DEDIFFERENTIATION AND TRANSDIFFERENTIATION
Stem cells are defined as cells that have self-renewing capacity and the ability to differentiate into multiple cell types[8]. Stem cells are present in mammalian embryos at the blastocyst stage as well as in the tissues and organs of adults. While embryonic stem cells (ESCs) have the ability to differentiate into any cell type present in the adult body, adult stem cells are capable of generating and replacing terminally differentiated cells in specific tissues and are involved in the continual maintenance and repair of tissues and organs throughout life[9,10].
An unidirectional developmental model suggesting that pluripotent stem cells progressively lose their pluripotency as they differentiate along developmental pathways until they reach a terminally differentiated state was widely accepted for several decades[11]. It was believed that adult stem cells have the ability to generate only the differentiated cell phenotypes of the tissue in which they reside. At the beginning of the 21st century, numerous studies challenged this strict hierarchy of stem cells and their unidirectional differentiation. The phenomenon of stem cell plasticity, or transdifferentiation, has emerged, suggesting that some adult stem cells have phenotypic potential that extends beyond the cell types of their resident tissue[12,13]. It was proposed that some stem cells under specific conditions might diverge from their predetermined pathway and generate cells of a different tissue by entering into a process of transdifferentiation, or that mature cells dedifferentiate into cells with a stem cell phenotype and eventually differentiate into cells of a different tissue[9].
In 1962, Gurdon[14] first proposed the hypothesis that the genome of every specialized cell of an adult organism has all the information required to develop into all different cell types. This hypothesis was proven in the seminal publication of Takahashi and Yamanaka[15], which demonstrated that adult differentiated cells could be reprogrammed into induced pluripotent stem cells that have the ability to differentiate into any of the endodermal, ectodermal and mesodermal cell lineages.This reprogramming is achieved by the overexpression of stem cell-associated genes,also known as stemness factors or Yamanaka factors, in the differentiated cells.
These findings validated the phenomenon of dedifferentiation and transdifferentiation, laying the groundwork for cancer stem cell (CSC) theory and the discovery of CSCs.
CSCs
The first indication of the existence of cancer cells with a stem cell phenotype came from the study of teratomas, which showed that undifferentiated cells preferably gave rise to nontumorigenic differentiated cells[16]. The CSC hypothesis was proposed in line with these data, suggesting that tumors comprise a mixture of malignant stem cells and their benign counterparts[17].
CSCs represent a small subpopulation of tumor cells with the capabilities of selfrenewal, differentiation, and tumorigenicity when transplanted into an animal host[18]. CSCs and stem cells share similar properties, including self-renewal ability,unlimited growth potential, invasiveness and blockade of differentiation (reviewed in[19]), whereby indefinite self-renewal capability enables CSCs to initiate and maintain tumor growth.
CSCs were first identified in acute myeloid leukemia[20] and subsequently in a wide variety of tumor types, including melanoma, osteosarcoma, leukemia, breast,colorectal, brain, prostate, pancreatic, ovarian, liver and lung cancer[21-23].
Advances in whole-genome sequencing have revealed the remarkable genetic complexity of malignant tumors and the presence of subpopulations of cells with distinct genotypes and phenotypes[24]. The distinct genotypes of cancer cells within the tumor endow the cells with different biological features and phenotypes,providing the basis for intra- and inter-tumor heterogeneity[25]. The CSC model explains this phenotypic and functional heterogeneity among cancer cells[26-29].
It has been proposed that CSCs originate from either adult tissue-resident stem cells or from differentiated cells that have been reprogrammed to a pluripotent state by the process of dedifferentiation[30]. The CSC hypothesis proposes that many heterogenic cancers are organized in a hierarchical fashion based on the differentiation capacity of the cells comprising the tumor[31]. It has been suggested that this hierarchical order recapitulates the normal tissue hierarchy established by healthy stem cells. Thus, CSCs generate cellular heterogeneity by imposing a differentiation hierarchy by generating a range of distinct cell types present within the tumor[26]. However, the established hierarchy is not permanent, and under specific conditions, could be reversed as terminally differentiated cells become dedifferentiated and regain CSC properties[22,27].
CSCs have been shown to exhibit high plasticity, resulting in changes in their phenotypic and functional appearance in response to chemo- and radiotherapeutics that cause alterations in the tumor microenvironment (TME)[30]. The negative effects of senescence can directly promote cancer stemness by increasing CSC plasticity,activating stemness pathways in non-CSCs, and promoting senescence escape and subsequent activation of a stemness pathway[30].
CSCs share a number of unique features that distinguish them from other tumor cells. CSCs are believed to be responsible for cancer initiation, progression, metastasis,recurrence and drug resistance[32]. Epithelial CSCs express many genes/pathways typically associated with normal stem cells (reviewed in[31]). In many types of tumors,some CSCs acquire epithelial-to-mesenchymal transition profiles through the upregulation of the expression of specific genes driving metastasis. CSCs are suggested to be responsible for drug resistance and cancer relapse due to their ability to self-renew and differentiate into heterogeneous lineages of cancer cells[33]. Drug resistance has been linked to the ability of CSCs to become quiescent, upregulate the expression of enzymes such as aldehyde dehydrogenase, and upregulate the expression of antiapoptotic proteins and multidrug resistance pumps that increase chemotherapeutic elimination from cells, resulting in low intracellular drug concentrations[31].
Accordingly, the majority of CSC features depend on the deregulation of signaling pathways that, in turn, rely on the altered activity of specific transcription factors(TFs).
SOX TFs AND CSCs
SOX (Sry-related HMG box) proteins constitute a large family of diverse and wellconserved TFs comprising at least 20 SOX family members in mammals[34]. They have been divided into eight distinct groups designated A-H based on their structure,expression profiles and homology (Table 1)[35]. The SOXB group is further subdivided into subgroup B1 comprising SOX1, SOX2 and SOX3 and subgroup B2 consisting of SOX14 and SOX21[36].

Table 1 Classification of the human SOX genes[35]
SOX proteins display properties of both classical TFs and architectural components of chromatin (reviewed in[37]). SOX TFs possess a 79 amino acid HMG domain that enables their specific DNA binding and additional domains involved in transcriptional regulation (reviewed in[37]). SOX TFs exert regulatory functions to activate or repress gene transcription through specific interactions with their partner factor(s) and by establishing contacts with the basic transcription machinery[38].
Several essential roles have been attributed to SOX TFs since their discovery. SOX TFs are a component of a regulatory network and, together with other TFs, signaling pathways, epigenetic modifiers and microRNAs, govern diverse cellular processes during development, such as the maintenance of stem cell pluripotency, cell proliferation, cell fate decisions, germ layer formation and the terminal differentiation of cells into tissues and organs (reviewed in[39,40]). However, the roles of SOX TFs are not limited to development as they also influence cell survival, regeneration and death, and control homeostasis in adult tissues[41,42].
Numerous studies have reported the roles of SOX TFs in the preservation of stem cell characteristics, playing a part of regulatory network required to establish ESCs and to maintain their pluripotent and proliferative state. SOX2, OCT4 (octamerbinding TF 4) and NANOG (named after the mythological Celtic land of the everyoung, “Tir nan Og”)[43] comprise the core transcriptional circuit that orchestrates the maintenance of stem cell self-renewal and pluripotency[44].
Accumulating evidence has demonstrated that OCT4, SOX2 and NANOG are the core factors in a pluripotency gene network involved in the induction, maintenance and loss of pluripotency (reviewed in[45,46]). The state of pluripotency, displaying regulatory flexibility, is supported by a highly interconnected pluripotency gene regulatory network that functionally relies on a set of core pluripotency TFs. The state of pluripotency integrates external signals and exerts control over the decision between self-renewal and differentiation at the transcriptional, post-transcriptional and epigenetic levels[46]. Growing evidence shows that the overexpression of these core stemness-associated TFs occurs in various types of human cancers, and aberrant expression of these TFs is associated with tumor initiation, progression and therapy resistance (reviewed in[47]). Although core stemness-associated TFs play critical roles in maintaining pluripotency and self-renewal in both ESCs and CSCs, distinct mechanistic functions between them have been proposed[48].
Since the first reviews of the involvement of theSOXgenes in cancer[49], the roles of particular members of this gene family in the development of multiple cancer types have been extensively studied. It has been shown thatSOXgenes may act as oncogenes, tumor suppressor genes, or both depending on the cellular context(reviewed in[50,51]). Oncogenic SOX TFs are overexpressed in multiple cancer types,exerting their oncogenic functionviaseveral mechanisms, including the promotion of proliferation, suppression of apoptosis, promotion of metastasis, and maintenance of CSCs[50,52-54].
SOX2 TF is one of the most studied SOX proteins, and during the last decade,aberrant SOX2 expression has been associated with various types of cancer (reviewed in[55,56]). In various tumor types, SOX2 has been linked to cancer stemness, and elevated SOX2 expression has also been associated with chemotherapy resistance[57],endothelial-to mesenchymal transition[58], promotion of clonogenicity, andin vivotumorigenicity[57,59]. In addition, elevated SOX2 expression in glioblastoma (GBM) is associated with increased cell motility and tumor spreading, and its expression is detected amongst circulating CSC islets[60-62]. A summary of the various roles that SOX2 plays in cancer is presented in Figure 2.
The impact of SOX proteins on brain tumors has been extensively studied in the last few years, and significant contributions have been made to the understanding of the roles of SOX in the initiation, progression, dedifferentiation and spreading of glioma tumors.
GBM
GBM, classified as a grade IV glioma tumor, is the most common and malignant primary brain tumor[63]. GBM accounts for more than 60% of all brain tumors in adults and approximately 45% of all gliomas; the annual incidence of GBM is 3-4 cases per 100000 people[64-66]. The ratio of GBM occurrence is, to some extent, higher in men than in women (1.6:1)[63]. The median overall survival of patients with this type of tumor is approximately 12-15 months[67], with a median age at diagnosis of 65 years[68]. It has been demonstrated that the 5-year survival rate of GBM is 5.3%[69]. It is the most aggressive malignant brain tumor in adults, is among the most vascularized solid tumors (reviewed in[70,71]), consists of heterogeneous cell populations in different phases of differentiation[72], and recapitulates most of the hallmarks of cancer, including uncontrolled proliferation, resistance to apoptosis,dysregulation of the cell cycle and angiogenesis (reviewed in[73]).
GBMs are defined as primary, arisingde novo, or secondary, developing from lowergrade glioma tumors (grade II or III astrocytoma) through malignant transformation[74]. Four clinically relevant subtypes of GBM have been identified based on molecular expression patterns: proneural, neural, classical and mesenchymal[75]. It has been demonstrated that various molecular subclasses can be detected within the same tumor[76]. It is reported in the literature that the proneural and neural GBM subtypes arise in or near the subventricular zone, while the mesenchymal and classical subtypes occur distal to the subventricular zone (reviewed in[77]). Furthermore, based on the gene expression profile, Parket al[78] identified three prognostic subtypes of GBM:mitotic (favorable), intermediate and invasive.
GBM persistently communicates with its TME, and the TME contributes to the tumorigenesis and progression of GBM (reviewed in[79,80]). The GBM TME is extremely heterogeneous, consisting of the extracellular matrix, tumor cells such as glioma stem cells (GSCs), and non-tumor cells, including endothelial cells, pericytes,microglia, immune cells, oligodendrocytes, neurons, astrocytes and myeloid-derived suppressor cells (reviewed in[81]). Extracellular vesicles containing soluble proteins,DNA, mRNA and noncoding RNAs enable communication between the GBM and the TME, and these vesicles contribute to angiogenesis, invasion, evasion of apoptosis and resistance to drugs (reviewed in[82,83]). Furthermore, it has been shown that GBM cells communicate by thin membrane channels (tunneling nanotubes), and the results obtained by Valdebenitoet al[84] revealed that these nanotubes mediate the protection of GBM cells from temozolomide (TMZ) and ionizing radiation treatment.
The precise cell of origin of GBM is still a controversial issue. It has been reported in the literature that neural stem cells (NSCs) of the subventricular zone and oligodendrocyte precursor cells represent two major candidates for the GBM cell of origin (Figure 3) (reviewed in[85]). Furthermore, several studies proposed that GBM might arise from the malignant transformation of glial and astrocyte precursor cells as well as from the dedifferentiation of astrocytes and neurons (reviewed in[85-87]).

Figure 3 Potential cells of origin of glioblastoma. During the neural differentiation process (black arrows), neural stem cells differentiate into fate-restricted precursors that are capable of differentiating into neurons, astrocytes or oligodendrocytes. Glioblastoma cells can arise due to the accumulation of cancer driver mutations in different cell types or due to the dedifferentiation of mature cells (orange arrows). Modified from[224]. References are included in the main text.
The current standard therapeutic treatment of GBM includes tumor resection by surgery, followed by radiotherapy and concomitant chemotherapy with TMZ[67].However, GBM still remains incurable and usually recurs due to a high level of intertumoral and intratumoral heterogeneity demonstrated at the histological, cellular and molecular levels (reviewed in[88-90]), the presence of GSCs, infiltration into the healthy brain parenchyma, the high rate of migration from the tumor core and the ability to generate secondary tumors (reviewed in[91]).
GBM tumor initiation, progression, relapse, and resistance to treatments are associated with GSCs (reviewed in[92,93]), and these cells represent one of the main therapeutic targets.
ROLE OF SOX GENES IN GBM AND CORRELATIONS WITH PROGNOSIS AND SURVIVAL OF PATIENTS
Almost all members of theSOXgene family contribute to the malignant phenotype of GBM by regulating cell proliferation, migration, invasion, apoptosis, stemness, tumorigenicity or angiogenesis. The correlation ofSOXgene expression levels with patient clinical outcomes has been documented (Table 2).

Table 2 SOX gene activity in glioblastoma and correlation with the clinical outcome of glioblastoma patients
VariousSOXgenes exert oncogenic properties in GBM.SOX1regulates the selfrenewal and proliferation of patient-derived cells, tumor initiation and progression[94]. The expression ofSOX1in GBM is higher than that in the normal brain and lowgrade glioma and correlates with shorter overall survival[94].SOX2is one of the most studiedSOXgenes and is overexpressed in tumor samples from patients with GBM and in GBM cell lines[95-97].SOX2gene exerts oncogenic activity in glioma cells[98].High expression levels of SOX2, together with other stem cell markers, are observed in poorly differentiated GBM and are associated with its aggressiveness and poor prognosis[99-101]. It has been shown that recurrence of the tumor arises at the original site with a more invasive, aggressive phenotype and that is less sensitive to radiochemotherapy, and exhibits increasedSOX2expression levels compared to corresponding observations in the primary tumors[102]. In contrast, there are data showing thatSOX2expression is decreased in recurrent gliomas in comparison with its expression in primary gliomas and that patients with lowerSOX2expression have a poor prognosis after receiving chemoradiotherapy[103]. Furthermore, we have shown thatSOX3promotes the malignant behavior of GBM cells.SOX3affects GBM cell viability, proliferation, migration, invasion, and autophagy, and its high expression does not correlate with the overall survival of patients with GBM[104]. However,another study revealed that higherSOX3expression levels in glioma and glioma cell lines correlate with poor outcome[105].SOX9exhibits oncogenic functions; itssilencing reduces proliferative capacity, arrests the cell cycle in the G2/M phase and increases glioma cell apoptosis[106]. TheSOX9gene is also involved in the survival,proliferation and senescence of glioma cells[107]. Compared to the survival time of patients with lowerSOX9expression, the survival of glioma patients with increasedSOX9gene expression is shorter[106,108]. In addition,SOX10is expressed in gliomas[109] and, together with platelet-derived growth factor B, induces glioma development[110]. Moreover, high expression ofSOX10correlates with reduced overall survival[111].
Despite the well-established oncogenic activities of someSOXgenes, others show tumor suppressive properties and are associated with good clinical outcomes. It was shown thatSOX5expression was high in glioma samples and glioma cell lines compared to theSOX5expression in the normal adult brain and that the survival of patients with GBM with IgG antibodies against SOX5 in the sera was significantly prolonged compared to the survival of patients with no IgG antibodies[112].Furthermore,SOX5overexpression inhibits clone formation of human glioma cells[113]. Schlierfet al[114] showed that the expression level ofSOX5in gliomas was the same or lower than that in adult brain tissue. In contrast, Tianet al[115] revealed that high expression ofSOX5correlates with poor prognosis in patients with GBM. These contradictory data probably arose as a consequence of GBM heterogeneity.Furthermore, it was shown that lowSOX6expression in patients with GBM contributes to a better survival rate than that in patients who display highSOX6expression[116]. Functional analyses have shown thatSOX7acts as a tumor suppressor. It has been demonstrated thatSOX7represses glioma cell proliferation and migrationin vitroand suppresses tumor formationin vivo[117]. It has been reported thatSOX11is overexpressed in glioma samples[118]. Hideet al[119] showed that downregulation ofSOX11expression is associated with a decrease in the survival of patients with GBM.Another study showed that SOX11, together with neurogenin 2, can reprogram glioma cells into neuron-like cells, inhibiting tumor growth and improving survival[120].Recent data confirmed the tumor suppressor activity of theSOX15gene in glioma tumors. Specifically,SOX15overexpression inhibits proliferation and invasion, while its upregulation delays tumor formationin vivo[121]. Furthermore, it has been demonstrated thatSOX21acts as a tumor suppressor[122] that inhibitsSOX2expression, induces apoptosis in human glioma cells[123] and leads to the differentiation of glioma cells, thus inhibiting glioma progressionin vivo[124]. Moreover, it has been shown that simultaneous ectopic expression of theSOX5,SOX6andSOX21genes in human primary GBM cells induces senescence and apoptosis, thus reducing their malignant potential[125]. Their downregulation increases the capacity of stem cells from the subventricular zone of the mouse brain to form glioma tumors upon encountering oncogenic stimuli[125].
Angiogenesis represents one of the hallmarks of GBM[126]. Upregulation ofSOX7expression promotes tumor angiogenesis in a mouse model of high-grade glioma, and its high expression in GBM patients is associated with poor survival and early recurrence[127].SOX13silencing reduced the tube formation abilities of U87 gliomaexposed endothelial cells[128]. Downregulation of SOX17 expression inhibited tumor angiogenesis in a mouse model of high-grade glioma, while no correlation between its expression level and the clinical outcome of patients with GBM was documented[127].
SomeSOXgenes display both oncogenic and tumor suppressive functions depending on the cellular context. For instance,SOX4exerts tumor suppressor activity in glioma cells by inducing GO/G1 cell cycle arrest and inhibiting growth[129]. In contrast, this gene is involved in maintaining the stemness of glioma-initiating cells[130]. There are also contradictory data regarding the correlation betweenSOX4expression and the clinical outcome in patients with GBM. Galatroet al[131] reported that patients with GBM with combined high expression ofID4,SOX4andOCT4exhibited lower survival. On the other hand, Zhanget al[129] demonstrated thatSOX4expression is positively correlated with a good prognosis for patients with primary GBM.
Taken together, these reported findings demonstrate that almost allSOXgenes are expressed in GBM and the expression levels of the majority of these genes are associated with the prognosis and survival of patients with GBM.SOXgenes that exert oncogenic roles are usually associated with a poor prognosis and shorter survival,making them potential candidates as prognostic biomarkers and future therapeutic targets.
GSCs
GSCs were among the first CSCs identified in solid tumors[132]. GSCs are one of the major contributors to GBM heterogeneity; these cells exhibit a high proliferation rate and self-renewal capacity (reviewed in[71]), an ability to differentiate into diverse cell types, the ability to recapitulate a whole tumor (reviewed in[133]), enhanced invasive capacity, increased angiogenic potential[134] and an ability to initiate tumor growth and progression shortly after the surgical removal of the primary GBM tumor(reviewed in[92]). GSCs usually express specific markers, such as CD133 (Prominin 1),CD44, NANOG, SOX2, OCT4, POU class 3 homeobox 2 (POU3F2), MYC protooncogene (c-Myc), Spalt Like TF 2 (SALL2) and KAT8 regulatory NSL complex subunit 2[135,136]. It has been reported that GBM cells are able to interconvert between GSC and non-GSC states[137]. Additionally, it has been found that a single GBM tumor contains different subsets of GSCs: proneural and mesenchymal[138].
Numerous studies have demonstrated the importance of signaling pathways in the maintenance of GSC properties, including the Hedgehog (HH)[139], Wnt/β-catenin[140], NOTCH[141], Epidermal growth factor receptor[142], Phosphatidylinositol-3-phosphate kinase/AKT/Mammalian target of rapamycin[143], Mitogen-activated protein kinase (MAPK)[144], Inhibitory kappa B kinase/Nuclear factor-kappa B (NFκB)[145], Transforming growth factor-beta (TGF-β)/Small mothers against decapentaplegic[130] and Janus kinase/Signal transducers and activators of transcription (STAT)[146] pathways (Figure 4). It has been proposed that the regulation of the GSC phenotype results from a complex interplay between multiple signaling cascades[143,147-149]. Accordingly, targeting several GSC-related signaling pathways at the same time seems to be necessary for the increased efficiency of anticancer treatments. A variety of small-molecule inhibitors of the HH, NOTCH and WNT/β-catenin signaling pathways have been identified/developed, along with inhibitors that target different pathways simultaneously, and the efficacy of some of these inhibitors have been demonstrated in preclinical and clinical studies[150,151].
GSCs are enriched in recurrent gliomas (reviewed in[152]), and they form more aggressive and diffuse tumors after intracranial transplantation in athymic nude mice than GSCs originating from the primary tumor of the same patient[153]. These factors make GSCs a primary therapeutic target. Choet al[154] described five methods for targeting GSCs: development of GSC-specific chemotherapeutic agents, application of radiosensitizers, usage of immune cells capable of attacking GSCs, induction of GSC differentiation and gene therapy.
GSCs reside in the perivascular and perinecrotic GBM niches (reviewed in[79]).Perivascular niches contain normal and reactive astrocytes, pericytes, gliomaassociated microglia/macrophages, myeloid cells, fibroblasts and normal NSCs in addition to GSCs and tumor cells (reviewed in[79]). GSCs residing in the perivascular niches divide slowly, are resistant to therapy and produce high levels of proangiogenic factors (reviewed in[155,156]). It has been reported that endothelial cells stimulate GSC self-renewal and tumorigenicity; on the other hand, GSCs may regulate and contribute to the tumor vasculature by producing cytokines and chemokines and by transdifferentiating into endothelial cells or pericytes (reviewed in[157]). Perinecrotic niches contain GSCs around necrotic foci induced by hypoxia (reviewed in[158]). Hypoxia induces signaling pathways that can impact GSC self-renewal, proliferation and invasion (reviewed in[158]). Cells expressing molecular markers of both hypoxia and GSCs (e.g., SOX2, NANOG, CD133) are largely found in perinecrotic niches (reviewed in[134]).
SOX GENES AND MAINTENANCE OF GSCs PROPERTIES
It has been reported thatSOXgenes play important roles in the maintenance of GSC properties (Figure 5). Knockdown ofSOX1expression in GSCs impairs their selfrenewal, proliferation, viability and tumor-forming capacity, whileSOX1overexpression promotes their malignant phenotype[94]. Among the SOX TF family, SOX2 is considered to be the most important player in the maintenance of GSC properties,including their tumorigenicity. Gangemiet al[159] demonstrated that silencing theSOX2gene in GSCs terminated cell proliferation, consequentially leading to the loss of tumorigenicity. The results obtained by Songet al[160] indicate that SOX2 plays a primary role in the regulation of CD133-positive GBM cell tumorigenicity. On the other hand, Coxet al[161] demonstrated that a 3-fold increase inSOX2 expression in GBM cells decreases their capacity to proliferate and generate spheres. These results suggest that the level ofSOX2expression must be precisely regulated by tumor cells during gliomagenesis. Furthermore, it has been found that co-expression of SOX2,oligodendrocyte TF 2 (OLIG2) and zinc finger E-box binding homeobox 2 is involved in the maintenance of GSCs[162]. Downregulation of SOX2 expression impairs sphereforming capacity, self-renewal, viability, proliferation, migratory and invasive capabilities of brain tumor stem cells derived from GBM[98]. Furthermore, knockdown of SOX3 expression in GSCs decreases the proliferation, migration and invasion capabilities of these cells and enhances their apoptosis[163], while knockdown of SOX4 expression in glioma-initiating cells results in reduced sphere-forming and selfrenewal capacities[130]. Silencing ofSOX9leads to inhibition of sphere-forming capacity[164], whileSOX9overexpression increases the sphere-forming capacity of GBM cells and induces the formation of larger tumors[165]. In glioma-initiating cells,inhibition of the midkine/anaplastic lymphoma kinase axis promotes SOX9 degradation, resulting in a decrease in the self-renewal and tumorigenic capacities of glioma-initiating cells[166]. Kurtsdotteret al[125] demonstrated that simultaneous forced expression of SOX5, SOX6 and SOX21 factors blocks the tumor-inducing capacity of human primary GBM cells.
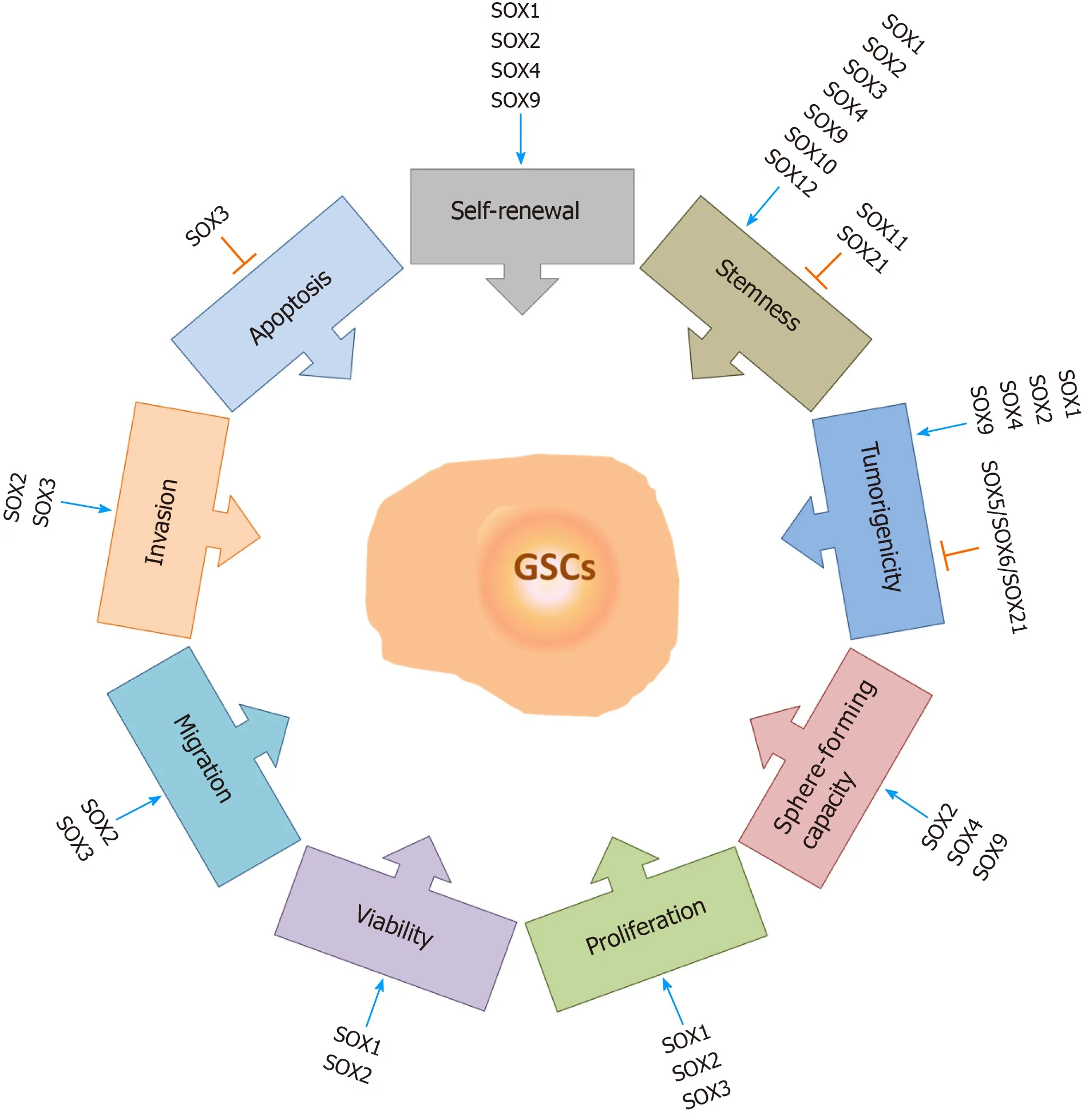
Figure 5 The roles of SOX transcription factors in glioma stem cells. SOX transcription factors exert numerous functions in glioma stem cells (GSCs).They influence GSC self-renewal, stemness, tumorigenicity, sphere-forming capacity, proliferation rate, viability, migration and invasion capacities, and apoptosis.Some of the members of the SOX gene family are involved in the resistance of GSCs to therapy. Blue arrows represent stimulation, and orange bars represent inhibition. References are included in the main text. GSCs: Glioma stem cells.
THE ROLES OF SOX GENES IN STEMNESS AND DIFFERENTIATION OF GSCs
It has been reported that SOX TFs have important functions in both the maintenance of the stemness of GSCs and the dedifferentiation of glioma cells (Figure 6). The results obtained by Berezovskyet al[100] indicate thatSOX2may play a central role in the regulation of dedifferentiation and acquisition of GSC properties. They found that knockdown ofSOX2expression blocks the dedifferentiation of HF2303 GBM cells and reduces their tumorigenicity. Additionally, downregulation ofSOX2expression in brain tumor stem cells derived from GBM led to an increase in the expression of differentiation markers[98]. Furthermore, it was shown that the TGF-β/SOX4/SOX2 pathway plays a crucial role in the maintenance of stemness and tumorigenicity in glioma-initiating cells[130]. It has been shown that the combination of SOX2, POU3F2,SALL2 and OLIG2, detected in the proneural subtype of GBMs, is able to reprogram differentiated tumor cells into GSCs[136].
In addition to the overwhelming evidence of the importance of theSOX2gene in the maintenance of the stem cell properties of GSCs, it was shown that the other two members of the SOXB1 subgroup also play important roles. Dedifferentiation of GBM cell lines is accompanied by an increase inSOX1expression. Additionally,SOX1expression was higher in patient-derived GSCs than in conventional glioma cell lines,and after the differentiation of these cells, the levels of SOX1 decreased dramatically[94]. Recently, we revealed thatSOX3expression was higher in patient-derived GSCs,as well as in oncospheres derived from GBM cell lines, compared toSOX3expression in their differentiated counterparts, suggesting thatSOX3expression is associated with the undifferentiated state of GBM cells[104].
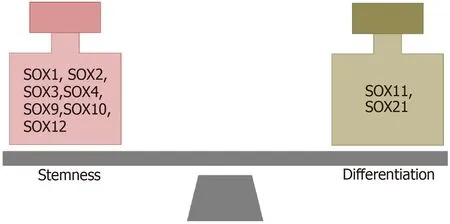
Figure 6 SOX transcription factors involved in the maintenance of stemness and differentiation of glioma stem cells. References are included in the main text.
Additionally, the roles of otherSOXgenes in the regulation of stemness and differentiation of GSCs have been described. It was found that the number of differentiated cells is increased after SOX4 knockdown in glioma-initiating cells[130]. On the other hand, the expression ofSOX9is increased in oncospheres derived from GBM cell lines compared to its expression in their differentiated counterparts, and silencing ofSOX9downregulated the expression of the stem cell markers CD133, NESTIN and SOX2[164]. Overexpression ofSOX10in LN229 GBM cells increased the expression levels of the stemness markers CD133, NESTIN, OCT4, NANOG and CD44[111]. On the other hand,SOX11overexpression in glioma-initiating cell-like cells and human gliomainitiating cells from malignant glioma blocked their tumorigenic ability by inducing neuronal differentiation[119]. The results obtained by Wuet al[167] indicate that SOX12 regulates the stemness of GBM cells and that SOX2, CD133, and OCT4 act as downstream effectors of SOX12 in these cells. Additionally, the results obtained by Caglayanet al[124] suggest thatSOX21overexpression decreases the stem-like cell properties of glioma cells and initiates their differentiationin vivo.
Together, these reported findings indicate that SOX TFs play important roles in GBM and GSCs. Since GSCs are the main drivers of GBM progression and therapy resistance, further studies are needed to comprehensively delineate the roles of SOX TFs in these cells.
SOX GENES AND CHEMO-AND RADIORESISTANCE OF GSCs
GBM inevitably recurs following the initial therapy as a result of intratumoral heterogeneity and resistance to chemo- and radiotherapy, which are considered to be promoted by GSCs (reviewed in[133,156]). Conventional chemo- and radiotherapies affect the proliferative cells within the tumor, while GSCs successfully evade their effects through multiple intrinsic GSC features and adaptive mechanisms (Figure 7).These mechanisms include enhanced expression of the DNA mismatch repair geneMGMT(O6-methylguanine-DNA methyltransferase)[168], high expression of drug efflux transporters[168], activation of the antiapoptotic pathway[168], increased replication stress leading to a constitutively active DNA damage response[169,170]and aberrant activation of multiple signaling pathways, such as Notch, HH, Wnt/βcatenin and NF-κB[171-174].
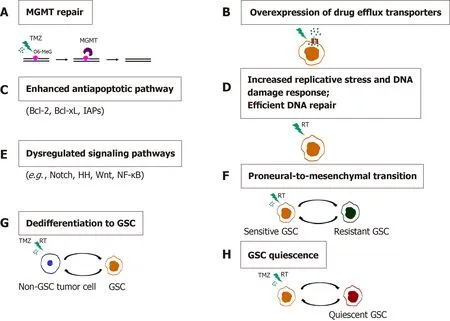
Figure 7 Mechanisms of chemo- and radioresistance of glioma stem cells. A: O6-methylguanine-DNA methyltransferase (MGMT) repair; B:Overexpression of drug efflux transporters; C: Enhanced antiapoptotic pathway; D: Increased replicative stress and DNA damage response. Efficient DNA repair; E:Dysregulated signaling pathways; F: Proneural-to-mesenchymal transition; G: Dedifferentiation to GSC; H: Glioma stem cells (GSC) quiescence. Resistance of GSCs to temozolomide is achieved through increased expression of MGMT, drug efflux transporters and antiapoptotic proteins. Replicative stress, constitutively active DNA damage response and efficient DNA repair confer radioresistance to GSCs. Aberrantly activated signaling pathways, the ability of non-GSC tumor cells to dedifferentiate, proneural-to-mesenchymal transition and the capacity of GSCs to become quiescent contribute to the chemo- and radioresistance of glioblastoma.References are included in the main text. RT: radiation; O6-MeG: O-6-methylguanine; GSC: Glioma stem cell; TMZ: Temozolomide.
One of the essential features of cancer cells that confers resistance to therapy is their plasticity,i.e., the capability of going through phenotypic changes in response to different environmental signals without genomic alteration. It has been shown that extrinsic factors such as radiation[175] or TNF-α (tumor necrosis factor α) through activation of the NF-κB pathway[171] promote proneural to mesenchymal shift of GSCs leading to a radioresistant phenotype[171]. Segermanet al[176] demonstrated that GBMs contain a heterogeneous population of glioma-initiating cells that exhibit variable levels of resistance to radiation and drugs[176]. The observed variations in therapy responses are the result of the slow shifts between the proneural- and mesenchymal-like cell states regulated by the changes in DNA methylation of the cisregulatory regions of mesenchymal master regulator genes.
Another important mechanism underlying GBM resistance to therapy is the ability of differentiated cancer cells to undergo dedifferentiation into cancer stem-like cells.Auffingeret al[177] showed that long-term exposure of glioma cells to clinically relevant doses of TMZ caused the phenotypic switch of non-GSCs towards GSCs that exhibited expression of pluripotency- and stemness-related markers CD133, SOX2,OCT4, and Nestin, a highly infiltrative phenotype and increased chemoresistance.Analyses of the long-term effects of sub-toxic doses of radiation on patient-derived differentiated GBM cells and the phenotypic and molecular changes in GBM towards stemnessin vitrowere demonstrated, including the expression of stem cell markers SOX2, Nestin and OLIG2, as well as increased tumorigenicityin vivo[178].
Quiescent, slow-cycling CSCs play a critical role in tumor recurrence, as they escape the effects of chemo- and radiotherapy that mostly eradicate proliferative cells in the tumor bulk. TMZ treatment in a transgenic mouse model of glioma demonstrated that recurrent tumors derived from dormant glioma cells exhibited stem cell properties[179]. Qaziet al[180] revealed that the combined chemoradiotherapy of primary human GBMs resulted in acquired resistance of brain tumor stem cells, which was accompanied by increased expression of stemness-associated factors SOX2 and BMI1 and an enhanced self-renewal capacity of primary GBM cells.
The roles of theSOX2andSOX9genes in the chemo- and radioresistance of GSCs have been well documented among the members of the SOX TF family (Figure 8).High expression ofSOX2in GBM patient-derived CD133-positive GSCs was shown to play a key role in promoting the self-renewal of GSCs, thereby contributing to their drug resistance[160]. Yanget al[181] showed that the miR-145/OCT4/SOX2 axis plays an important role in the chemoradioresistance of CD133-positive GSCs. Inhibition of stemness factors SOX2 and OCT4 by miR-145 was followed by an increased sensitivity of patient-derived CD133-positive GSCs to TMZ and radiation. The reduced chemoradioresistance of GSCs was also accompanied by a decrease in the expression of a few drug resistance (MDR1- multidrug resistance protein 1 andABCG2-ABC subfamily G member 2) and antiapoptotic genes (Bcl-2- B-cell lymphoma 2 andBcl-xL- B-cell lymphoma-extra large)[181]. Overexpression ofID-4(inhibitor of differentiation 4)induced the dedifferentiation of human glioma cells into glioma stem-like cells and downregulated the expression ofSOX2-repressing miR-9*, leading to enhancedSOX2expression[182]. SOX2, as a direct transcriptional regulator, upregulates the expression of ABCC3 and ABCC6 transporters, conferring chemoresistance on ID4-induced glioma stem-like cells and patient-derived GSCs[182]. Furthermore, it was revealed thatSOX2is a direct transcriptional target of Forkhead box M1 (FOXM1) and that the FOXM1/SOX2 axis promotes the stemness and radioresistance of GBM[183].

Figure 8 Schematic illustration of the involvement of SOX2 and SOX9 in the chemoresistance and radioresistance of glioma stem cells.The expression of stemness-related transcription factor SOX2 is elevated in glioma stem cells (GSCs) through various mechanisms (decreased expression of miR-145 and miR-9* that directly target SOX2, direct transcriptional activation by Forkhead box M1 and through mammalian target of rapamycin signaling) contributing to chemo- and radioresistance of GSCs. SOX2 directly activates the expression of ABCC3 and ABCC6 transporters. mTOR signaling affects the SOX2/SOX9 axis,contributing to chemoresistance. The ERK1/2/miR-124/SOX9 axis and direct targeting of PDK1 (pyruvate dehydrogenase kinase 1) by SOX9 have a role in resistance to radiation and temozolomide, respectively. References are included in the main text. GSCs: Glioma stem cells; FOXM1: Forkhead box M1; mTOR:Mammalian target of rapamycin; PDK1: Pyruvate dehydrogenase kinase 1.
A study by Garros-Regulezet al[165] demonstrated the correlation between high levels of SOX2 and SOX9 expression and resistance of GSCs to TMZ. The SOX2/SOX9 axis acts downstream of the mTOR signaling pathway and contributes to the observed chemoresistance. The authors proposed pharmacological inhibition of SOX2 by using specific inhibitors of the Sonic Hedgehog (SHH) and mTOR signaling pathways as a promising therapeutic approach to overcome TMZ resistance in a group of GBMs that display high expression levels of SOX2 and SOX9[165]. Wanget al[164] uncovered thePDK1(pyruvate dehydrogenase kinase 1) gene as a direct target of SOX9 that was shown to play a role in the regulation of GSC self-renewal and resistance to TMZ. The silencing ofSOX9and inhibition ofPDK1both reduced the activity of the AKT pathway and sensitized GSCs to TMZ treatmentin vivo, highlighting the SOX9/PDK1 axis as a new promising target for more efficient GBM therapy. Most recently,Sabelströmet al[184] demonstrated the critical role of the ERK1/2/miR-124/SOX9 axis in triggering the differentiation of stem-like GBM cells towards a neuronal phenotype.Inhibition of ERK1/2 activation by using a MEK inhibitor caused the enhanced expression of miR-124 and a reduction in the expression of its target gene,SOX9,consequently leading to decreased tumorigenicity and radioresistance in patientderived GBM xenografts. These results imply that the induction of neuronal differentiation of GSCs represents a promising therapeutic approach in anti-GBM therapy.
CONCLUSION
Combatting GBM has been a demanding and challenging goal for decades. Heterogeneity of cell types within the tumor, the contribution of patient-to-patient variability to the growth, the response to treatment driven by the genomics of each tumor and the transition between proliferative and non-proliferative phases make GBM extremely difficult to treat (reviewed in[185]). GBM is one of the deadliest cancers, and GBM studies face many challenges, some of which are unique to brain tumors (blood-brain barrier and immunosuppressive environment) and others that are shared with other tumors (tumor heterogeneity at the cellular and molecular levels, plasticity, and the presence of CSCs). One of the most essential hallmarks of GBM is tumor heterogeneity,which has been documented at the histological, cellular and molecular levels. Both intertumor heterogeneity (comprising distinct genetic alterations between different individual tumors) and intratumor heterogeneity (comprising diversity within individual tumors) have been identified in GBM (reviewed in[90]). The heterogeneity of GBM has been defined at multiple molecular levels using-omics technologies.Heterogeneity documented at the genomic and transcriptomic levels enables the molecular classification of histopathologically indistinguishable tumors into different subtypes. An advanced understanding of intratumor heterogeneity has been achieved by single-cell sequencing. Patelet al[186] performed the first single-cell RNA-seq and confirmed that GBM subtype classifiers are variably expressed across individual cells within a single tumor, while a recent study revealed that malignant cells in GBM exist in four main cellular states that are reminiscent of the canonical neurodevelopmental cell types[187]. Another level of heterogeneity is linked to treatment-induced plasticity and temporal heterogeneity. By sequencing exomes of initial low-grade gliomas and recurrent tumors from the same patients, Johnsonet al[188] revealed that at least half of the mutations in the initial tumor were undetected at recurrence in 43% of cases.Recently, a database of initial tumor and tumor recurrence samples from patients assembled by the Glass consortium revealed that 35 out of 222 patients exhibited treatment-related hypermutation at recurrence, while 70% of the cohort had an increased mutational burden after recurrence compared with the mutational burden of their initial tumor[189]. Taken together, these findings indicate that GBM heterogeneity provides additional layers of complexity that must be taken into consideration in the development of more effective therapies aimed at improving survival in GBM patients.
Remarkable progress has been made towards understanding the cellular and molecular biology of gliomas in the last decade. Advanced technologies have provided novel insights into multiple events that allow GSCs to acquire functional capabilities,enabling them to survive, proliferate and disseminate.
CSCs are considered to be, at least in part, responsible for drug resistance and cancer relapse due to their ability to self-renew and differentiate into heterogeneous lineages of cancer cells. Accordingly, the eradication of CSCs has become one of the major goals in cancer therapies. However, CSCs are very often therapy-resistant,representing the main obstacle to their eradication. Therapy resistance of CSCs is mediated by many different mechanisms, including the acquisition of dormancy,decreased apoptosis, increased DNA repair and drug efflux capacity, interaction with their supporting microenvironment within the CSC niche and hypoxic stability(reviewed in[190]). GBM tumors may contain different subsets of GSCs[138]. GSCs may adopt a quiescent state that protects them from chemotherapy and radiotherapy(reviewed in[191]), and these treatments might increase the population of GSCs over time (reviewed in[192]). GSCs successfully evade the effects of conventional chemoand radiotherapies through multiple intrinsic GSC features and adaptive mechanisms(Figure 7). GSCs enriched in recurrent gliomas acquire more aggressive and therapyresistant properties than GSCs originating from primary tumors that become more malignant and rapidly spread, leading to therapeutic failures[152,153,185].
All these properties make GSCs a promising but very complex and challenging therapeutic target for GBM treatment. A number of approaches for targeting CSCs have been developed, including inhibition of drug efflux ABC transporters, targeting signaling pathways, the microenvironment and cell surface markers of CSCs, differentiation therapy, immunotherapy and application of radiosensitizers (reviewed in[133,193-196]).
Dedifferentiation of tumor cells into a stem cell-like state is considered an additional source of cellular heterogeneity, pointing to tumor cell plasticity as an important driver of GBM heterogeneity[90]. Gimpleet al[137] proposed novel advanced therapies that are aimed at targeting tumor cell plasticity conferred by dedifferentiation(reviewed in[7,137]). One of the proposed approaches focuses on blocking dedifferentiation by applying a combination of therapies to prevent early resistance to therapeutics endowed by dedifferentiation and acquired lineage plasticity. Differentiation therapy is a novel approach that aims to target dedifferentiation by enhancing the conversion of dedifferentiated tumor cells towards permanently differentiated cells that are more sensitive to chemotherapy. The final proposed approach is focused on the differentiation of CSCs into harmless cell lineages that lack tumorigenic potency and metastatic potential by applying TFs or small molecules.
Significant conceptual and mechanistic similarities between cellular transformation in cancers and cellular reprogramming and dedifferentiation driven by various stem cell-associated pathways have been revealed[197]. The master regulators of stemness operate through transcriptional control of various stem cell-associated pathways during cellular reprogramming.
Since cell renewal is an ability that cancer cells must acquire to survive therapeutic treatment and reconstitute the tumor, master regulators of stemness have attracted significant attention in the treatment of GBM. SOX TFs are master regulators of stemness with known essential roles in GBM. Accordingly, SOX proteins and SOX2, in particular, have emerged as one of the major therapeutic targets for GBM.Overwhelming evidence of the importance of theSOX2gene in the maintenance of the stem cell properties of GSCs has been revealed. Numerous studies have demonstrated that glioma cells rely on SOX2 to maintain their tumorigenic activity, with GSCs displaying high levels of SOX2 (reviewed in[198]). SOX2 overexpression has been found in approximately 90% of human biopsies in GBM patient samples with enrichment in undifferentiated GSC populations. Downregulation of SOX2 in GSCs impairs cell proliferation and the ability of the cells to form tumorsin vivo.
It has also been shown that numerous SOX TFs are involved in the maintenance of the stemness of GSCs (SOX1, SOX2, SOX3, SOX4, SOX9, SOX10, SOX12) or play a role in the initiation of GSC differentiation (SOX11 and SOX21). Accordingly, the finetuning ofSOXgene expression levels is associated with the control of the balance between stemness and differentiation.
As outlined in this review, SOX TFs play multiple roles in GSCs. They influence GSC stemness, self-renewal, proliferation, differentiation, viability, migration,invasion, apoptosis, therapy resistance, sphere-forming capacity, and tumorigenicity(Figure 5). Therefore, innovative therapies aimed at targeting SOX TFs, and SOX2 in particular, are emerging as promising tools to combat GBM. These therapeutic approaches include the pharmacological inhibition of signaling pathways involvingSOXgenes[165,199,200], application of natural and synthetic compounds that control the expression of these genes[201-204], inhibition of SOX-DNA binding[205], inhibition of ubiquitylation and degradation of the SOX2 transcriptional repressor[206], immunotherapy[207,208], epigenetic silencing by miRNAs[181], blocking SOX function by peptide aptamers[209] and direct transcriptional repression ofSOXgene expression[210] (Figure 9).
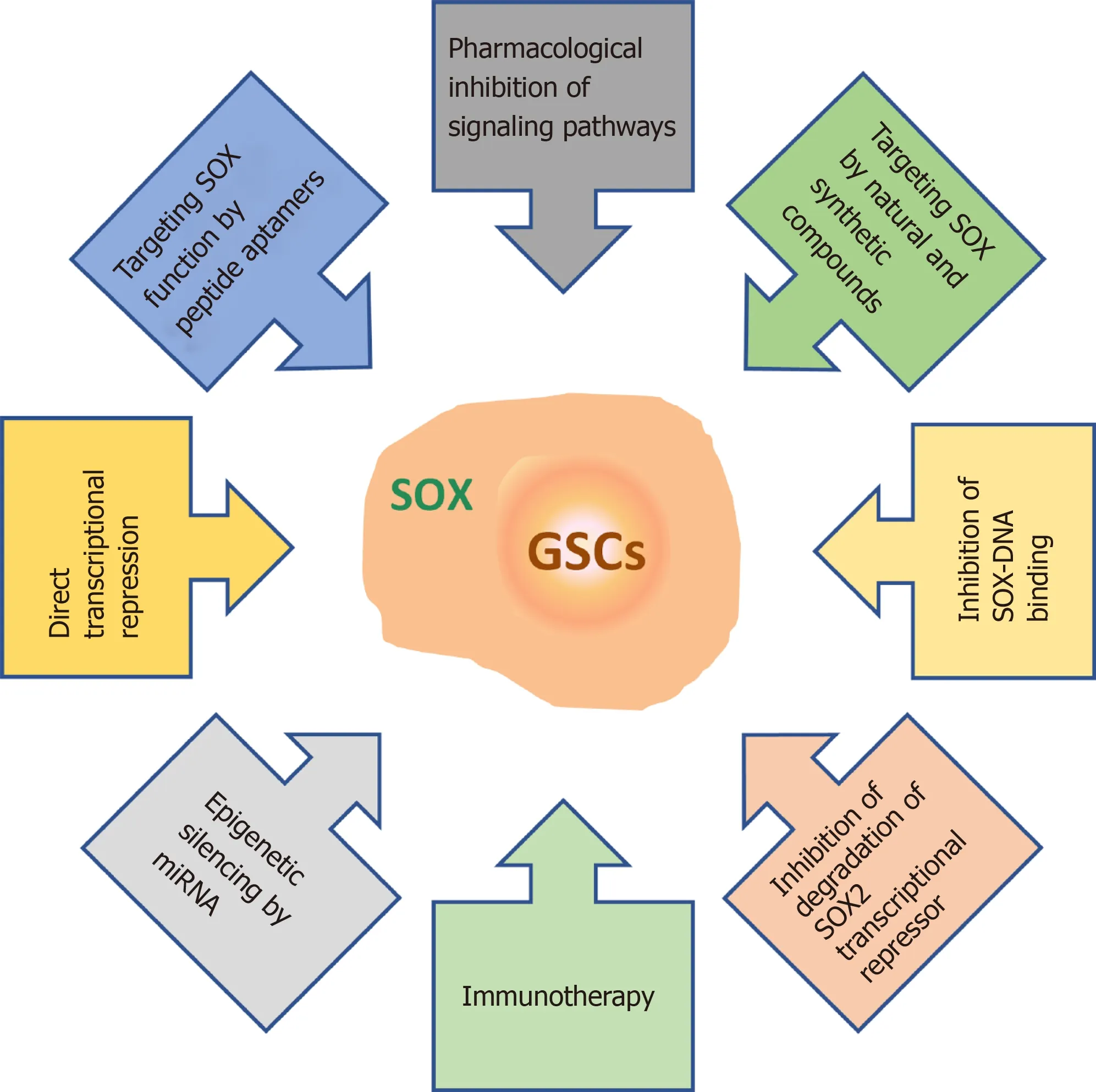
Figure 9 Overview of the therapeutic strategies for targeting SOX in glioma stem cells. References are included in the main text. GSCs: Glioma stem cells.
Modulation ofSOXgene expression in GSCs represents an attractive approach to combat GBM.SOXgene expression in GSCs might be targeted by applying different approaches. One of the approaches to modulateSOXgene expression is treatment with natural plant products. Studies on natural plant products have revealed that they can act as effective antitumor agents[211]. Forty-nine per cent of the small molecules approved for tumor treatment between 1940 and 2014 were natural plant products or their derivatives[211]. Some of these products have anti-GBM potential alone or in combination with other chemotherapeutics[211-214]. Our results revealed that extracts fromAnthrisci cerefoliiandOnonis spinosaL. plants have anti-GBM activity[215,216].Furthermore, it has been reported that nuciferine obtained from the leaves ofN.nuciferaGaertn. inhibited GBM progressionviathe SOX2/AKT/STAT3/Slug pathway[201]. On the other hand, it was found that curcumin, which inhibits the proliferation of glioma cells and induces apoptosis and autophagy, simultaneously induces glioma cells to become stem-like and enhances the expression of SOX2, SOX4 and OCT4[203].In addition to natural plant products, antibiotics such as salinomycin and actinomycin D reduce SOX2 expression in GSCs, indicating that drug repurposing may provide additional options for GBM treatment[202,204].
The expression ofSOXgenes in GSCs could be targeted by pharmacological inhibitors of signaling pathways that regulate GSC properties. Rapamycin, an inhibitor of mTOR complex 1, and cyclopamine, an inhibitor of the SHH pathway, reduced the expression of SOX2 and SOX9 in GBM cell lines[165]. Additionally,SOXgenes could be targeted by engineered artificial TFs that selectively modulateSOXgene expression. Stolzenburget al[210] developed zinc finger-based TFs that reducedSOX2expression, leading to reduced proliferation and colony formation in breast cancer cells.
Another approach is targeting SOX TFs in GSCs by the delivery of vectors that express miRNAs that were shown to specifically downregulate the expression of SOX proteins in GSCs. For example, the delivery of miRNA-145 to GBM patient-derived CSCsin vitroandin vivoresulted in the downregulation of the expression of stemness factors SOX2 and OCT4, suppression of tumorigenicity, increased sensitivity to TMZ and radiation and enhanced survival rate of xenotransplant mice when applied in combination with radiotherapy and TMZ[181].
GSCs could also be combated by vaccine approaches. It was demonstrated that vaccination of immunocompetent mice with Sox2 peptides significantly delayed tumor development[207]. Furthermore, Uedaet al[208] found that the SOX6 peptide induces SOX6 peptide-specific cytotoxic T lymphocytes, which are able to lyse GSCs derived from GBM.
In the last decade, immunotherapy that engages naturally occurring or genetically engineered oncolytic viruses has provided an additional strategy, especially for the treatment of cancers with poor prognosis, such as GBM. Although oncolytic viruses can infect both normal and cancer cells, they can only replicate in cancer cells[217].Preclinical and clinical trials have pointed to the capability of oncolytic viruses to stimulate antitumor immune responses by recruiting T cells (reviewed in[218,219]). It has been reported that the Zika virus exerts oncolytic activity against GSCs. Compared to Zika virus infection of differentiated GBM cells, the Zika virus preferentially infects and selectively kills GSCs and stem-like cells in a SOX2-dependent manner[220]. It has also been reported that an immunotherapeutic approach in gliomas that uses a vesicular stomatitis virus expressing HIF-2a, SOX10 and c-Myc together with checkpoint inhibitors anti-PD1 and anti-CTLA-4 enhances the antitumor response[221].
The current therapeutic strategies for GBM have not yet provided a cure and have only resulted in a slight improvement in patient survival. Considering that GBM therapies result in poor outcomes and that GBM treatment is one of the most expensive cancer treatmentper capitain the United States[222], there is a critical need for novel and more effective therapeutic options. Novel approaches to GBM treatment will require the fine-tuning of multimodal therapeutic strategies that simultaneously target numerous hallmarks of cancer cells to win the battle against GBM, one of the deadliest cancers.
杂志排行

World Journal of Stem Cells的其它文章
- Translational products of adipose tissue-derived mesenchymal stem cells: Bench to bedside applications
- Unveiling the morphogenetic code: A new path at the intersection of physical energies and chemical signaling
- Alternative RNA splicing in stem cells and cancer stem cells:Importance of transcript-based expression analysis
- Retina stem cells, hopes and obstacles
- Considerations for the clinical use of stem cells in genitourinary regenerative medicine
- Age and genotype dependent erythropoietin protection in COVID-19