药食同源植物甘葛藤的全长转录组分析
2021-11-01李向荣蔡时可王继华
梅 瑜,李向荣,蔡时可,顾 艳,王继华
(1.广东省农业科学院 作物研究所,广东省农作物遗传改良重点实验室,广东 广州 510640;2.汕尾市陆河县农业农村局,广东 汕尾 516600)
粉葛为豆科植物甘葛藤(Puerariathomsonii)的干燥根[1],性味甘、辛、凉,具有解肌退热、生津止渴、升阳止泻、通经活络、解酒毒等功效[2-3]。粉葛的药用功能始载于东汉时期的《神农本草经》及《名医别录》,此外,在古典药用植物《本草品汇精要》等书籍及现代的《中药大辞典》和《中国药典》中都有记载[4]。甘葛藤种质资源丰富,在我国主要分布在云南、四川、西藏、江西、海南、广东、广西等地,部分地区还获得了国家地理标志认证,如广东的合水粉葛、火山粉葛、竹山粉葛、庙南粉葛、鹤山粉葛和活道粉葛等[5]。作为我国传统的药食两用植物,其有效药用物质主要集中在根部,包括淀粉类、黄酮类、葛根苷类、香豆素类、三萜类和三萜皂苷类等100多种化合物,花中还鉴定出15种新的化合物[6]。其中,葛根素是功能研究较多的化合物,也称葛根黄素,是异黄酮类衍生物,在葛根药用成分中研究相对较多,具有退热、扩冠状的作用,在临床上常用于冠心病心绞痛、高血压的治疗[7]。
粉葛医药保健作用越来越受到人们的重视,开发出来的产品种类也越来越丰富,开发前景广阔,但其研究主要集中在化合物的分离和鉴定方面。转录组是基因功能研究的基础和出发点,连接基因组遗传信息与蛋白质组。单分子测序技术为第3代测序技术,是近年逐渐成熟的新一代测序技术,在转录组学研究方面具有独到优势。研究非模式植物的功能基因组学时,获得全长转录本可推进基础研究水平,第3代测序技术为此提供了较好的解决方案[8]。目前,全长转录组测序技术已经广泛应用于动植物的研究中,如利用全长转录组开发姜荷花(Curcumaalismatifolia)的分子标记[9];利用联合测序技术研究丹参(SalviamiltiorrhizaBunge)活性成分的生物合成及调控机制[10];利用三代测序PacBio平台获得苦苣菜(SonchusoleraceusL.)叶片全长转录组参考序列,结合BGISEQ-500平台的数字基因表达谱技术,分析不同浓度NaCl处理2 d苦苣菜叶片转录组表达谱,探究苦苣菜响应盐胁迫的主要基因及代谢途径[11],目前在粉葛的研究上还未见相关报道。本研究对甘葛藤进行全长转录组测序分析和基因注释,并对转录因子(Transcription factor TFs)、R基因和SSR标记进行鉴定,为甘葛藤优良品种选育和利用提供帮助。
1 材料和方法
1.1 试验材料
一年生的甘葛藤植株取于广东省广州市南沙区庙街。分别采集植株叶片、嫩茎和根,立即在液氮冷冻,存储于-80 ℃冰箱。
1.2 试验方法
1.2.1 RNA提取 采用植物RNA提取试剂盒(TIANGEN(天根生化科技北京)有限公司,货号DP441)分别提取甘葛藤3个器官的总RNA,用1%琼脂糖凝胶电泳法检测RNA的完整性,DNA Marker为全式金BM101(TransGen Biotech),15 000 bp。使用NanoPhotometer spectrophotometer和安捷伦2100生物分析仪(Agilent Technologies,Palo Alto,CA)评估RNA质量和完整性。选择3个OD260nm/230nm>1.8的总RNA等量混合,构建文库,用Pacbio三代测序仪(Pacific Biosciences,US)进行全长转录组测序。
1.2.2 建库测序与拼接组装 采用PacBio三代测序仪对甘葛藤的cDNA进行测序。检测合格后的RNA使用ClontechSMARTer PCR cDNA Synthesis Kit反转录为第1链cDNA;PCR扩增合成双链cDNA;使用AMPure PB Bead对PCR扩增产物进行纯化用于SMRTbell文库构建,包括DNA损伤修复、末端修复、连接Adapter、文库质量评估,将SMRTbell文库退火结合引物和聚合酶,采用MagBead Loading上机测序,由广州基迪奥生物技术有限公司完成。利用Fast QC评估原始数据,利用NGS QC工具包对数据进行过滤(去除含接头或含N的读长,大于10%未知核苷酸或大于40%低质量碱基的读长,Q值≤20),获得质量较高的读长,然后用Trinily软件进行转录本的拼接组装[12]。
1.2.3 单基因功能注释 利用BlastX程序对NCBI数据库(Nr)、Swiss Prot蛋白数据库、KEGG和COG/KOG数据库进行比对分析,用Blast2GO软件分析GO的功能注释和Nr注释结果[13],用WEGO软件进行功能分类[14]。使用EST扫描软件确认蛋白质编码序列和序列方向,将单基因的蛋白质编码序列与智能数据库(SMART database)比对(版本号SMART 06/08/2012),获得蛋白质结构域注释。
1.2.4 转录因子(TFs)和R基因分析 利用BlastP将单基因蛋白编码序列与植物转录因子数据库v4.0(http://planttfdb.cbi.pku.edu.cn/)进行比对,预测TF家族;通过BlastP将单基因的蛋白质编码序列与植物R基因数据库PRGdb(http://PRGdb.crg.eu/wiki/Main_Page)进行比对,预测甘葛藤中的R基因。
1.2.5 SSRs预测 依据配置参数使用软件MISA(http://pgrc.ipk-gatersleben.de/misa/)对甘葛藤转录组Isoform进行搜索,寻找Isoform中的SSR,并用Primer软件设计SSR侧翼区的引物。
2 结果与分析
2.1 甘葛藤全长转录组测序数据质控
利用单分子长读数测序技术(Single-molecule longread sequencing technology, SMRT),总共获得19 680 274 333 bp,去除低质量序列后,获得10 994 967个高质量reads,N50为 2 524 bp,平均长度为1 789 bp。Full Passes数目大于等于2的序列为参数,提取reads中的CCS序列得到492 223个reads,共1 177 436 362 bp,平均长度为2 392 bp。其中,含有5′primer的 CCS有444 921条,含有3′primer的CCS 有457 237条,含有polyA的CCS有449 010条。全长reads为416 045条,全长非嵌合序列(Full-length non-chimeric,FLNC)384 072条,占78.03%,共906 989 814 bp,平均长度2 361 bp;其次是非全长序列(Non-full-length reads)占15.29%;全长嵌合体序列(Full-length chimeric)和短序列(Short)占比较少,分别为6.50%,0.19%。
对reads进行聚类和校正,得到193 955条初步校正的一致性序列(Unpolished consensus isoforms);用Quiver算法对一致性序列进行进一步的校正得到高质量序列146 312条,低质量46 138条,平均长度2 365 bp。一致性序列进行去冗余得到高质量序列90 856条,共218 049 687 bp,最长序列为14 015 bp,最短为131 bp,平均为2 399.95 bp,N50为2 777 bp,GC含量为40.31%。
2.2 甘葛藤全长转录组功能注释
利用公共数据库对90 856个转录本进行比对注释,结果85 239个单基因被NR、Swissprot、KOG和KEGG数据库注释。其中,84 675个基因被注释到NR数据库,占93.2%;73 149个基因被注释到Swissprot数据库,占80.51%;被KOG数据库注释的基因有59 931个,占65.96%;KEGG数据库注释的基因有38 460个,占42.33%;未被以上数据库注释的基因有5 617个,占组装基因的6.12%,可能是新基因不能被注释;被四大数据库共同注释的基因有32 037个。
利用BlastX进行同源序列比对,结果发现,甘葛藤和大豆(Glycinemax)的单基因匹配率最高,共有39 603个(43.59%),其次是野大豆(Glycinesoja,12 598个,13.87%)、木豆(Cajanuscajan,10 230个,11.26%)、苜蓿(Medicagotruncatula,4 621个,5.09%)、绿豆(Vignaradiata,3 674个,4.04%)和赤豆(Vignaangularis,3 246个,3.57%)等(表1)。

表1 物种分布统计Tab.1 Statistical table of species distribution
2.2.1 KOG分析 共有59 931个基因被KOG注释分为25类(图1)。按照功能来分,大多数基因属于一般功能预测(20 168;22.20%),其次是信号转导机制(15 265;16.80%)和翻译后修饰、蛋白转换、伴侣(12 519;13.78%)等。少数单基因被分配到细胞动力,仅有81个;其次是细胞外结构和核结构,分别为362,445个;另有4 243个基因未知功能。有2 891个基因与次生代谢物生物合成、运输和分解代谢相关,这些基因可能对甘葛藤中次生代谢物的特定生物合成、运输和积累有重要作用。
2.2.2 GO 功能注释分类 30 837个基因被注释到GO数据库,被分为三大类:生物过程、细胞组分和分子功能。46.93%的基因分布在生物过程,其中代谢过程的基因最多(19 087,21.01%),其次是细胞过程(17 710,19.49%)、单有机体过程(13 326,14.67%)、生物调节(5 678,6.25%)。30.89%的基因分布在细胞组分中,细胞(11 388,12.53%)和细胞部分(11 387,12.53%)是本亚类的最主要的组分,其次为细胞器(9 183,10.11%)、膜(6 139,6.76%)和膜部分(4 559,5.02%)等。22.18%的基因被注释到分子功能,催化活性(18 249,20.09%)最为突出,其次是结合(15 027,16.54%)、运输活性(1 516,1.67%)等(图2)。
2.2.3 KEGG通路分析 通过KEGG数据库比对分析,共有22 330个基因被注释到132条途径。其中代谢途径(9 368,41.95%)分布的基因最多,其次是次级代谢产物生物合成(5 220,23.48%)、抗生素生物合成(2 372,10.62%)、不同环境的微生物代谢(2 258,10.11%)、碳代谢(1 783,7.98%)、氨基酸生物合成(1 261,5.65%)等(表2)。
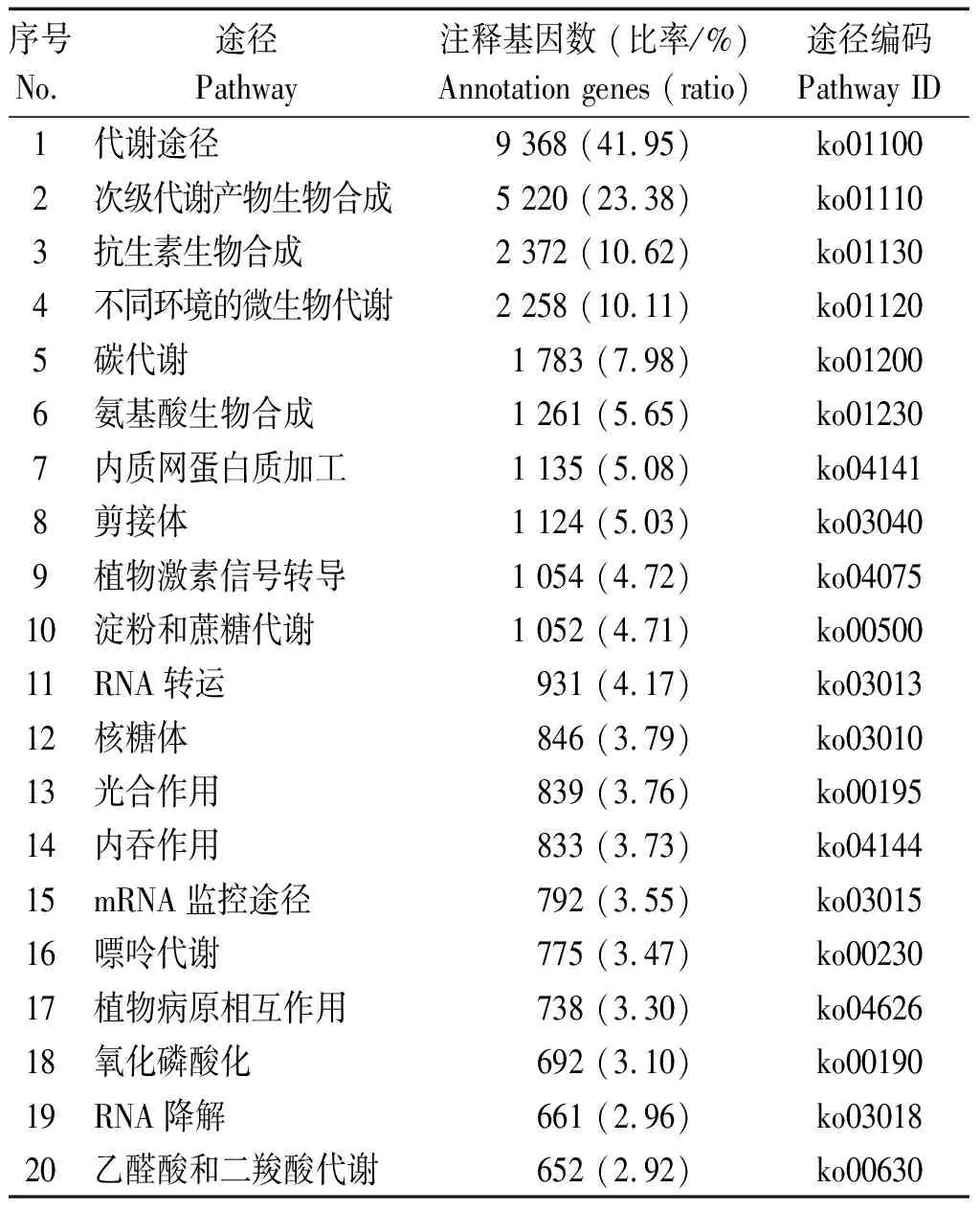
表2 KEGG通路富集Tab.2 KEGG pathway enrichment
2.2.4 黄酮类生物合成基因的鉴定 黄酮类是植物的苯丙氨酸代谢过程中产生的植物次生代谢产物,是一类由肉桂酰辅酶A侧链延长后环化形成以苯色酮环为基础的酚类化合物。异黄酮类物质是粉葛的主要活性成分,其中如葛根素、鸢尾苷元和鸢尾苷,是粉葛的主要的药用成分之一[15]。根据粉葛的转录组数据绘制其黄酮类生物合成代谢途径(图3)。在苯丙酸生物合成途径中,苯丙氨酸为底物,由苯丙氨酸解氨酶(Phenylalanine ammonialyase,PAL)将氨基基团从苯丙氨酸分子中移除而形成了肉桂酸(Cinnamic acid);然后在4-香豆酸-CoA连接酶(4-coumarate-CoA ligase,4CL)和肉桂酸-4羟化酶(Cinnamic acid 4-hydroxylase,C4H/CYP73A)作用下,在肉桂酸分子中添加上一个羟基基团而形成对-香豆酸(p-Coumarate);其侧链结合辅酶A分子(CoA),从而转变为对-香豆酸辅酶A分子(p-Coumarate-CoA),作为黄酮类化合物合成的起始底物。对-香豆酰辅酶A在查尔酮合酶(Chalcone synthase,CHS)的催化下产生柚皮素查尔酮或异甘草素,经查尔酮异构酶(Chalcone isomerase,CHI)作用,分别形成柚皮素或甘草素;之后,柚皮素作为中间产物进入代谢途径,甘草素作为底物进入异黄酮生物合成途径。除此之外,对-香豆酰辅酶A在莽草酸O-羟基肉桂酰基转移酶(Shikimic acid O-hydroxy cinnamoyltransferas,HCT)、5-O-(4-香豆蔻酰基)-D-奎宁酸3′-单加氧酶(5-O-(4-coumaroyl)-D-quinate 3′-monooxygenase,C3′H)作用下形成咖啡酰-CoA,经查尔酮合酶催化形成2′,3,4,4′,6′五羟基查尔酮(2′,3,4,4′,6′Pentahydroxy chalcome);或经咖啡酰辅酶A O-甲基转移酶 (Caffeoyl-CoA O-methyltransferase,CCoAOMT)催化形成阿魏酰辅酶A,再由查尔酮合酶催化形成4,2′,4′,6′,四羟基-3-甲氧基查尔酮(4,2′,4′,6′,-Tetrahydroxy-3-methoxy chalcome)。甘葛藤黄酮类的合成在不同酶的催化下参与复杂的代谢过程,产生的次生代谢产物较多。在甘葛藤转录组数据中,与黄酮类合成相关的基因110个,其中,26个编码HCT,3个编码CHS,7个编码CHI(表3)。
2.2.5 转录组中TFs和R基因分析 TFs在生物过程的基因表达模式主要起调节作用,如黄酮类化合物的生物合成途径[16]。根据比对结果,3 507个基因被划分到55个不同的转录因子家族(图4)。其中Basic/Helix-Loop-Helix(bHLH)转录因子类的基因最多(290个),其次是C2H2(249个)、TALE(225个)、C3H(221个)、WRKY(179个)、bZIP(173个)等,最少的是GRF(1)和NZZ/SPL(1),这些转录因子信息为甘葛藤次生代谢产物的生物合成和抗逆性研究提供了依据。
植物R基因在识别病原菌的特异无毒性(Avr)基因和刺激诱导抗病的信号转导级联中起着关键作用[17]。14 127个基因被分为17个不同的R基因类别。受体样蛋白(RLP)的种类最多(3 633个),其次是TNL(2 729个)、N(2 239个)、NL(1 621个)、CNL(1 203个)等(图5);分配到PTO(1个)的基因最少,其次是RLP-Malectin(9个)和RLK-Malectin(2个)。
2.2.6 转录组中SSRs标记位点的分布 SSR标记被认为是基于DNA多态性的一种标记技术,是进行遗传多样性检测和遗传图谱构建的有效工具之一[18]。从90 856个基因中筛选出了33 660个SSR,其中含有一个以上SSR的序列有6 628个,SSR以复合形式存在的序列有3 580个;在检测到的核苷酸基序重复序列中,二核苷酸(14 778个,43.93%)和三核苷酸(12 927,38.41%)占比较多,其次为四核苷酸(2 791,8.29%)、五核苷酸(1 550,4.60%)、六核苷酸(1 614,4.80%)。类型最丰富的是AG/CT(8 863,26.33%),其次为AT/AT(3 479,10.34%)、AC/GT(2 433,7.23%)和AAG/CTT(2 784,8.27%)(图6)。在此基础上,利用Primer Premier设计引物,为甘葛藤遗传多样性研究和遗传图谱构建提供了数据基础。
3 讨论
本草基因组学近几年的发展加快了药用植物基因资源的保护和利用。转录组分析是鉴定植物转录调控最为有效、准确和低成本的工具之一,也是本草基因组学研究的重要手段之一。在非模式植物的功能基因组学研究中,全长转录本提供的信息丰富,获得能够有力地推进相应基础研究水平,第三代测序技术为此提供了较好的解决方案。有助于揭示其生长发育、逆境应答机制和次生代谢产物的富集调控机制。目前,全长转录组测序已经广泛应用于动植物和微生物测序研究中,尤其是对于没有完成基因组序列测序的大多数药用植物,将三代高通量测序用于植物全长转录组研究,对具有生物活性次级代谢物的调控基因的探索和分子标记的开发具有重要意义[10]。如在药用模式植物丹参的研究中,基于二代测序获得的大部分序列不具有全长cDNA特性,而利用RNA-seq、Iso-SeqNGS和SMRT技术对丹参周皮、韧皮部、木质部进行转录本测序,获得了高质量的全长转录本,为丹参研究提供了完整的转录信息[8,19]。植物和传统药用植物中广泛存在着植物天然化合物,天然产物一直以来都是研究的热点。甘葛藤作为“药食同源”植物,黄酮类化合物是甘葛藤的主要活性物质之一,具有抗炎、抗氧化、保护心脏、降血糖等功效[20],越来越受到人们的青睐。但相对于其他农作物而言,甘葛藤的仍采用传统育种手段、比较落后,且育种周期较长、选育的品种药效难以保证。而且随着甘葛藤化学成分及药理作用研究的不断深入,其转录组学和基因组学的研究报道甚少,这不利于甘葛藤次生代谢产物合成途径的解析。本研究利用三代高通量测序平台对甘葛藤的3个不同组织进行混合测序,组装出90 856个单基因,有85 239个得到注释,获得完整的参考转录组。目前还缺乏甘葛藤的参考基因组,但是基于转录开发的SSR有助于改善甘葛藤的分子标记。同时,这些单基因序列和注释将有助于甘葛藤中新基因的发现和功能基因组分析。豆科有650个属,18 000种植物,葛属植物有20多种,本研究可为豆科植物的分子生物学研究提供参考。此外,葛根资源的系统收集和评价,以及新品种的培育是今后研究的重点,可为葛产业健康可持续发展提供保障。
