浅谈药品质量标准起草技巧和策略
2021-10-28韩新荣徐伯林夏振华
韩新荣 徐伯林 夏振华
【摘要】药物的安全性、有效性是建立在药物质量可控性的基础上,质量可控性是保证药品安全性和有效性的前提,也是研发和生产达到安全性和可控性的桥梁,在实际工作中首先保证质量可控性,质量可控性包括内容很多比如工艺稳定性、产品均一性、取样代表性、产品质量非边缘性等等,其中质量标准是一个很重要的保证产品质量可控性重要的技术标准,是保证药物安全性和有效性的重要一环。在文中,主要就对药品质量标准起草进行了探讨。
【关键词】药品质量;标准;起草技巧
【中图分类号】R95 【文献标识码】A 【DOI】
目前ICH[1]、欧州EDQM[2]、美国FDA[3]对质量标准制定了相关的技术指导性文件,国家食品药品监督管理局参照上述技术指导原则,于2005年发布了《化学药物质量标准建立的规范化过程技术指导原则》,但这些指导原则比较宽泛,本文就企业在实际制定质量标准过程中出现的一些纠结的问题进行厘清。
1 质量标准的分类
质量标准按照质量标准的目的可以分为货架期质量标准、放行质量标准、稳定性质量标准三种,货架期质量标准是药品在放行后至效期内应该执行的质量标准,也是药品的法定遵循的标准,包括药典标准,企业注册的药品质量标准等。
稳定性质量标准顾名思义即为稳定性考察所起草的质量标准,其项目可以比货架期标准项目少,减少的贮存中不会增加的项目,如残渣、重金属;或在贮存中会可能减少的项目,如残留溶剂;其项目可以比放行的项目多,增加的主要用于稳定性考察的研究性项目如晶型、溶出曲线、部分剂型的水分等项目,最后是否列入货架期标准根据稳定性数据来决定,对于微生物等样品量很大的项目可以在关键时间进行测定如加速6个月、长期12个月、24个月等。
2 制定质量标准策略
2.1 关于性状
外观是对药品的色泽和外表感观的规定,而性状范围更广,包括外观、臭、味,溶解度、吸湿性、晶型以及物理常数等,两者都是物理的而非化学指标,除外观颜色外,其他一般均为研究性参考性指标,可在工艺验证或产品研究时研究,不需要进行日常放行测定。
2.2 关于鉴别
鉴别方法分为专属性方法和证实性方法,鉴别必须含有专属性方法,如红外分光光度法、高效液相色谱法,同时鼓励使用证实性方法,紫外分光光度法由于没有专属性,只能作为证实性的鉴定方法,鉴别试验也不能太灵敏,应避免由限度内的杂质引起的错误的反应,对于盐类药物,除了鉴定活性成分还需要鉴定盐类基团。化学反应如颜色反应、沉淀反应、火焰反应是一个简单又科学的鉴定手段,在起草质量标准尽量使用,制剂由于辅料干扰,一般不建议采用红外分光光度法进行。
2.3 关于水分
水分是一个特殊项目,不能作为杂质来控制。因为水有两重性(结晶水是“好”水,对药物稳定性有利,游离水是“坏”水,对药物稳定性可能不利),而且水本身几乎没有毒性。
2.4 关于酸碱度
酸碱度有三种测定方法,如指示剂法(也称酸度或碱度)、标准酸碱法、pH法。使用何种方式,具体问题具体分析,如纯化水由于其不稳定使用指示剂法,而注射用水则使用pH法。
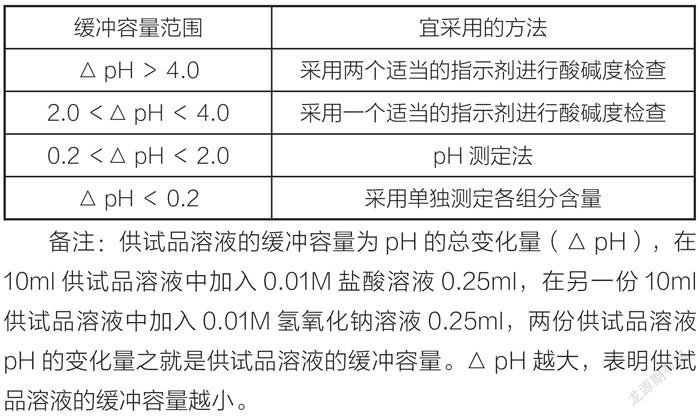
(1)对于易溶于水的含有強酸成盐的原料药,一般采用pH法控制,不再进行强酸的测定,如盐酸林可霉素,不再测定氯离子的含量,因为pH法更为灵敏。对于含弱酸盐类原料药或药物不溶于水或药物结构有酸碱基团,不宜采用pH进行质量控制,而是使用如HPLC测定弱酸根如富马酸喹硫平,总之,采用何种方式,首先是药物是否溶于水,不溶解的产品不宜使用pH,其次是使用药物在0.01mol/l的酸和碱中的pH差值(△pH)来决定,详见下表[2]。
备注:供试品溶液的缓冲容量为pH的总变化量(△pH),在10ml供试品溶液中加入0.01M盐酸溶液0.25ml,在另一份10ml供试品溶液中加入0.01M氢氧化钠溶液0.25ml,两份供试品溶液pH的变化量之就是供试品溶液的缓冲容量。△pH越大,表明供试品溶液的缓冲容量越小。
(2)测定一般使用pH计进行,目前市场有大量老款pH和新型的pH两种,为此,中国药典2020版由一种测定方法增加到两种测定方法,这是兼顾实际,与时俱进理念的体现,一种方法为三点校正,对斜率和漂移值制定要求,一种方法为两点校正,由于两点成一线,所以该法没有斜率要求,试验表明,这两种测定pH方法实际结果较为一致。
(3)pH值与温度有关,特别碱性溶液影响更大些,电极的氯化钾的浓度影响pH稳定性,需要特别注意氢氧化钙标准缓冲液特别不稳定,存放时应防止空气中二氧化碳进入,一旦出现浑浊,应弃去重配,如在只需测量大致pH值的情况下,可采用指示剂法或试纸法。
(4)pH除于温度有关外,还与浓度关,所以质量标准特别是放行报告单的pH指标需要标明温度和浓度,如25℃,50mg/ml水溶液。
2.5 关于溶液澄清度和颜色
溶液澄清度和颜色主要针对用于注射剂的原料药才需进行质量控制,如果产品本身有颜色,一般不控制颜色或控制其色级范围,如果产品本身无色,一般控制颜色小于相应色号,如1号黄色。澄清度测定注意放置位置,建议在散光灯上端与眼睛视线平行位置按照中国药典要求在1000LX下进行,如果和标准浊度液相近,需要左右调换位置进行判断。
2.6 关于晶型
同一物质具有两种或两种以上的空间排列和晶胞参数,形成多种晶型的现象称为多晶现象(polymorphism),晶型的质量控制非常重要,尤其稳定差的或难溶性并用于口服制剂多晶型的原料药,由于晶型影响药物的稳定性和溶解度,从而影响制剂的稳定性和疗效,在药品研发时需要认真对待,并通过稳定性考察药物在贮存过程中是否有晶型转变,如有,在质量标准中加以控制甚至需要进行各类晶型的定量测定,常见的晶型测定有X-射线衍射法、红外吸收光谱法、热分析法、偏光显微镜法、核磁共振法等方法,需要具体情况具体选择。
2.7 关于有关物质
杂质通常分为两种,一种是内源性杂质即与药品的生产工艺包括使用的物料及可能接触的设备有关的杂质,内源性杂质是通过制定质量标准进行质量控制的;另外一种外源性杂质,这个主要是污染或交叉污染产生的,外源性杂质是通过GMP来控制,无法通过技术手段进行控制。有关物质是内源性杂质的一种,是与工艺相关的物质,并且在HPLC或GC分析图谱中中出现杂质,测定有关物质共有外标法、面积归一化法、自身对照法、自身对照法加校正因子法、主成分外标法、主成分外标法加校正因子法,对于原料药和制剂,建议使用自身对照法加校正因子法或主成分外标法加校正因子法。
2.8 关于残留溶剂
2.8.1对于1类溶剂:生产工艺中应尽量避免使用,如工艺需要,被用作起始物料,应对其残留进行常规控制,可以是在适当的中间体中,也可以是在原料药成品中控制。如其存在于另一溶剂中(例如含苯的甲苯或丙酮),以下两种情况可以不进行常规检测:(1)已经采用一个验证过的检验方法证明1类溶剂在适当的中间体或原料药成品中不超过规定限度的30%,此时应提供连续6个中试批次或连续3个商业放大批次的支持性数据。(2)源溶剂中设定了1类溶剂的限度,评估后,该限度可以控制所用原料药中的该1类溶剂残留水平低于指南设定的水平(评估必须考虑干燥工艺中两种溶剂的挥发性)。
2.8.2对于2类溶剂:如被用作起始物料,应进行常规控制,根据其所用的合成步骤,可以是在适当的中间体中,也可以是在原料药成品中进行控制。如最后合成步骤以及精制使用的,应对原料药成品进行常规控制。
2.9 关于含量
2.9.1对于原料药:如采用非专属性的含量测定方法(如滴定法),除另有原因外,含量限度通常为98.0%-101.0%,但对于采用色谱技术(如液相或气相色谱法)含量测定方法,考虑实际误差,含量限度的上限一般为102.0%,制定101.0%不太科学,这点在制定质量标准时需加以注意。
2.9.2对于制剂:考虑到辅料的干扰,一般均采用专属性好的色谱法,标示含量一般为90.0%-110.0%,但由于现代生产设备和技术水平的提高,为了更好的保证药品的安全性和有效性,建議会结合原研的质量标准要求提高,如95.0%-105.0%,这个需在研究时要提前考虑,避免发补时出现麻烦。
3 制定质量标准的注意事项
3.1 关于药品名称
原料药稀释不用注明API名称,如:“取本品,加流动相溶解并稀释制成每lml中约含××mg的溶液,作为供试品溶液;…”;但制剂因有辅料必须注明,如:“装量差异项下的内容物,混合均匀,精密称取适量,加流动相溶解并稀释制成每1ml中含氯唑西林××mg的溶液,滤过,取续滤液作为供试品溶液;…”。
3.2 关于系统适用性误区
系统适用性溶液、对照溶液和对照品溶液等问题。样品溶液不能作为系统适用性溶液,有预检嫌疑,但经过氧化等降解处理可以作为系统适用性溶液测定分离度等系统适用性参数,此外,对照溶液和对照品溶液也有所不同,对照溶液一般为自身对照法测定杂质由样品溶液稀释而成,一般不可以作为系统适用性溶液进行系统适用性重复性测定,需另采用对照品溶液进行。
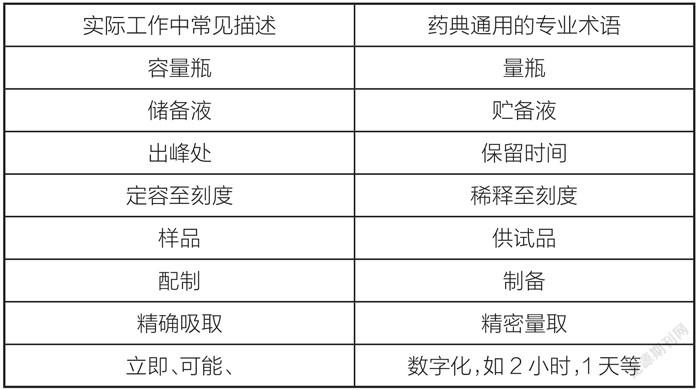
3.3 关于常见的用词纠正
3.4 关于溶液的稀释倍数
稀释倍数一般不作规定,1步稀释误差小,但消耗溶剂量大,论文[4]具体描述了各类稀释带来的误差,在制定时根据误差要求具体选择并通过方法验证进行,建议含量测定稀释次数不超过2步[2],有关物质等微量测定稀释次数尽量不超过3步。
3.5 关于分析色谱柱
现在色谱柱的品牌和种类特别多,药典方法考虑到商业避嫌,一般不注明具体品牌和型号,所以实际应用需要根据验证或确认来选择,非药典的方法可以注明色谱柱品牌。效能相当除考虑柱长、粒径大小外,还要考虑含碳量和比表面积等参数。通常含碳量低/比表面积大,有利于组分保留。
3.6 其他问题
3.6.1“与”和“和”的区别:连续加多种试液时,二种试液间用“与”,多种试液间用“、”最后两种试液间用“与”,不采用“和”与“及”。有关物质:用“和”,如:供试品溶液和对照品溶液。
3.6.2“用”与“加”:操作定量稀释或稀释至刻度均采用“用”字,如用××稀释至刻度,而不用“加”。
3.6.3注意质量标准项目顺序:顺序参照药典格式,不要自行改变,特别检查项,如制剂检查项次序分别为有关物质、溶出度、含量均匀度。
参考文献:
[1]ICH.Q6A:Specification:Test Procedures and Acceptance Criteria for New Drug Substances and Products:Chemical Substances[ S] .1999.
[2]European Directorate for the Quality of Medicines & Health Care欧洲药典质量标准的起草技术指南Technical Guide for the Elaboration of Monographs.4 th 2005.
[3]FDA.Guidance for Industry:INDs for Phase2 and Phase3 Studies Chemistry, Manufacturing, and Controls Information[ S] .2003.
[4]R.B.Lam,T.L.Isenhour.通过玻璃仪器的选择降低标准溶液制备过程中的相对误差. Analytical Chemistry, 1980, 53, 1158-1161.
