Effect of external electric field on crystalline structure and dielectric properties of Bi1.5MgNb1.5O7 thin films∗
2021-10-28ZhongzheLiu刘钟喆LibinGao高莉彬KexinLiang梁可欣ZhenFang方针HongweiChen陈宏伟andJihuaZhang张继华
Zhongzhe Liu(刘钟喆), Libin Gao(高莉彬), Kexin Liang(梁可欣), Zhen Fang(方针),Hongwei Chen(陈宏伟), and Jihua Zhang(张继华)
State Key Laboratory of Electronic Thin Films and Integrated Devices;School of Electronic Science and Engineering,University of Electronic Science and Technology of China,Chengdu 610054,China
Keywords: cubic pyrochlore,dielectric properties,tunability,DFT calculations
1. Introduction
Low-loss,high-kand dielectric tunable materials have attracted much attention for their huge potential in potable communication device application.[1–8]In these kinds of materials, barium strontium titanate (BaxSr1−xTiO3,BST) has been intensively studied for many years as dielectric tunable thin film materials. BST thin films have a high dielectric constant and large tunability.[9–14]However, relatively high dielectric loss is its main drawback to further applications. Many efforts have been made to improve its dielectric loss such as dopant,annealing and surface plasma treatment. These methods did not bring significant improvement to the dielectric loss because, as the high dielectric loss is induced by ferroelectric polarization, the dopant will also compromise the high tunability of BST materials. As the counterpart of BST materials, pyrochlore oxide materials show great potential and attract much interest for its moderate dielectric constant and tunability. Compared to the BST materials,the non-ferroelectric properties endow pyrochlore oxide materials with low intrinsic loss. Pyrochlore oxide materials have a general formula of A2B2O7, or A2B2O6O′with more structure details included. The bismuth-based cubic pyrochlore materials such as[Bi1.5Mg0.5][Nb1.5Mg0.5]O7(BMN)has been widely studied because of its high dielectric constant and low dielectric loss. It is reported that the dielectric constant and dielectric loss shows excellent stability to frequency.[15]Its dielectric tunability under bias electric field also broadens its application in microwave communication.[16–20]
Past studies mostly focused on the experiment of manufacturing process and method of preparing materials. For now, only a few works were focused on the theoretical studies. In these studies, the relationship between had been discussed mostly. Until recently,there was still a lack of knowledge about how doped atoms change the dielectric properties of the bismuth-based ceramics.
In this work,we prepared BMN ceramic with solid state reaction process. X-ray diffraction (XRD) analysis indicated that BMN ceramics were well crystallized and shows cubic pyrochlore structure. The structure of BMN model that used in density functional theory (DFT) calculations has the same XRD pattern that fits the experiment result. Raman spectra were performed to examine the vibration modes. We applied DFT calculations to study the dielectric property and calculate the Born effective charges and phonon frequencies and eigenvalues. The DFT Berry phase calculations’ results give out the polarization change before and after the electric field was applied. The results of theoretical calculations can derive the dielectric constant and intrinsic loss of the ideal materials.Comparing to the output result of experiment, we can find a way how to modified the materials properly.
2. Experiment procedure&calculation details
The cubic pyrochlore BMN ceramics are prepared using conventional solid-state reaction,Bi2O3,MgO and Nb2O5are prepared as starting materials. Samples were first crashed and mixed in stoichiometry. Then they are milled in a planetary ball mill for 12 hours with alcohol added as dispersant and lubricant. After milling procedure, the product was heated in a drying oven at 85°C atmosphere to expel alcohol from sample. Then the slurry was held in an aluminum crucible with a lid to pre-sinter at a temperature of 900°C for 2 hours. Another ball mill was conducted to join 2 wt%polyvinyl alcohol binders in the pre-sintering product for better sharping ability,deionized water is served as the dispersant rather than ethanol,and the ball mill procedure was endured for 12 hours. This time the powders are dried using a spray granulator to separate the solid from liquid. The dried and separated powders are compressed at a pressure of 20 MPa to build round-padshaped sample. Then we heat the round pad ceramic samples in a furnace at 600°C for 6 hours to expel the binders and then the temperature rises to 1120°C for 1 hour to get a well crystallized ceramic sample. Another procedure of constructing silver electrodes for the electrical contact is needed. At the end of all these processes, both sides of the ceramic pad are covered with silver paste and sintered at 680°C.
The crystalline structure was examined by x-ray diffraction analysis. And dielectric frequency spectra were measured by an Agilent 4294A precision impedance analyzer with a test oscillation voltage of 500 mV.[21]
The DFT calculations were carried out using the Quantum Espresso package.[22,23]The original structure was downloaded from Materials Project with its Zn cations replaced with Mg. A few studies reported a series of pyrochlore structures with similar formula Bi1.5XNb1.5O7(Xfor Mg,Cu,Ni,etc.),after replacing atoms we ran a series of relaxation calculations and obtained a more reasonable structure of BMN materials using the SG15 optimized norm-conserving Vanderbilt pseudopotentials.[24]In those pseudopotentials Bi(5d,6s,6p),O(2s,2p), Nb(4s,4p,4d,5s), Mg(2s,2p,3s) orbitals were included as valence electrons. Calculations were performed within PBE exchange-correlation functional. We ran a series of orthogonal single point tests to determine the cutoff energy andK-points. The cutoff energy was well tested and converged before relaxation and it ended up with a value of 85 Ry.TheK-points were generated by the Monkhorst–Pack method of a 2×2×2k-mesh. Two irreduciblek-points were left after the crystal symmetry was applied. To eliminate the internal forces induced by atoms replacing, we performed a series of variable-cell relaxations and fixed-cell relaxations to retain the max force under 10−6a.u./°A and total energy difference under 10−8Ry.
3. Results and discussion
The A2B2O7pyrochlore structure also can be described as the formula B2O6·A2O′,which are two interpenetrating networks in pyrochlore: [BO6] octahedra vertices form a threedimensional network. The octahedral B2O6network consists of{222}sheets of octahedra,sharing corners to form six and ternary rings,these sheets are often referred to as“hexagonal tungsten bronze”(HTB)layers. The O′and A atoms are filled in the large cavities between the BO6diamond net. These two kinds of atoms are also forming the second cuprite-like A2O′tetrahedral structure with four A atoms on the corner of the tetrahedron and the O′atom on the center. Each A atom is a part of two A2O′tetrahedrons and served as a connecting point. So A2O′tetrahedrons were linked and form a stable A2O′network. On the perspective of A cations, they are surrounded by six O atoms in the octahedral framework,which form a puckered hexagonal ring in the HTB layer,and two more O′atoms make the O′–A–O′link perpendicular to the puckered ring. The cubic pyrochlore adopts theFd3m(No. 227) symmetry and have four special atomic positions:A-16c,B-16d,O-48f and O′-8b.
We performed the XRD spectrum to examine the crystallization in BMN ceramic samples. The samples were scanned using CuKαradiation at a scan rate of 1°2/min, and the 2θangle ranges from 10°to 70°x-ray diffraction of BMN ceramics powder is shown in Fig. 1(a). The sharp and fully developed peaks evidently indicate the ceramics example is well crystallized and have a cubic pyrochlore structure. With no distortion, the second phase was detected in the XRD result. The intensity of(222)peak is the highest in all the peaks,which indicates that (222) plane is the preferred structure.[3]The lattice constant of well crystallized BMN ceramic calculated from XRD pattern can be derived using Bragg’s formula.The interplanar spacing of(222)plane isd222=3.060 °A.And the lattice constant is 10.60 °A, which shows good agreement with the DFT result of 10.58 °A(19.998 Bohr). It also proves partial replacement of both A-site cations and B-site cations by a smaller atom(such as Mg2+)leads to no shrinkage in cell parameters and remains the same cubic structure.
The scanning electron microscope(SEM)picture of polished and etched BMN ceramic and BMN thin film are provided in Fig. 1(b). the ceramic grains are closely contacted with its neighbor and thus formed tightly packed grains. No visible defect and blowhole can be seen in the SEM pictures.The average grain size of BMN ceramic is about 6.5µm. The small grain size and well contacted morphological characteristic indicate all the starting materials has been crashed and mixed properly and well sintered in the furnace.
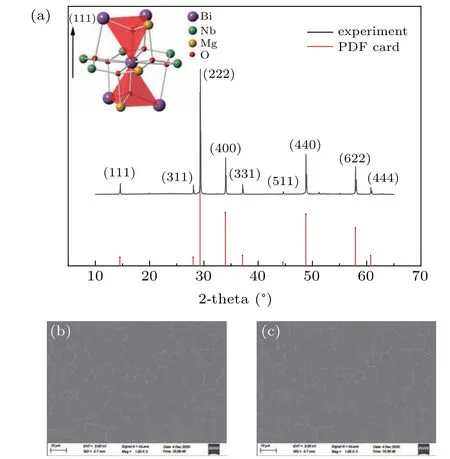
Fig. 1. (a) XRD of BMN ceramics and diffraction data from standard PDF card,(b)SEM images of BMN ceramic and(c)BMN thin film on sapphire.
The BMN thin film was deposited on the sapphire substrate using BMN ceramic as the sputtering target.In Fig.1(c),the SEM pictures shows nanoparticles of BMN ceramics have accumulated on the surface of the sapphire, and the average size of nanoparticles is~40 nm.
The group theory analysis of the ideal pyrochlore is conducted by Ronald[25,26]and the cubic pyrochlore structure consists of 26 normal modes
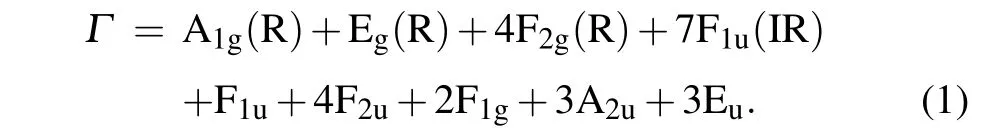

Table 1. Raman mode and assignment of BMN.
Among all the normal modes only one A1g, Egand four F2gmodes are Raman active, seven F1umodes are infrared(IR) active. F1umode is the acoustic mode. The rest modes are inactive. The BMN pyrochlore have all the Bi cations occupied A-site positions and Nb cations occupied B-site positions, the Mg cations are evenly filled in the rest positions of A- and B-sites. With both sites partially occupied by two types of atoms, the regular A2O′tetrahedron and BO6octahedra will distort and the center atoms will deviate from the original atomic position due to the difference in atomic radius and electronegativity between Mg and Nb(Bi).
The change in A–O′, A–O and B–O bond will affect the vibrational modes. The Raman spectra experience split degenerate modes and frequency shift as a consequence. The Raman spectra of the BMN ceramic are provided in Fig. 2 and more peaks were observed as a result of A-site atomic displacement. It has been reported that the A site cations and the O′anions contribute significantly in the displacement from the high-symmetry positions in the bismuth-based cubic pyrochlore structure. The DFT results of BMN cubic pyrochlores’ Raman peaks are also shown in Fig. 2 in the red bar. The cubic pyrochlore is the most stable phase at room temperature, but the cubic structure tends to transform into the other phase to reach a lower energy level below 100 K,some study reported that they found three infrared active imaginary modes in cubic pyrochlore vibration spectrum in DFT calculations.[27]We also found three imaginary modes with infrared activity in the BMN vibration spectrum given by DFT calculations. Compared to the conclusion given by the previous work, this result remained solid. The distortion of A2O′tetrahedron also has an influence on the BO6octahedra due to the twining binary network, although the BO6octahedra retains its inside structure relatively. The Raman mode assignment has been reported by several researchers and it is given in Table 1,The Raman assignment of the cubic pyrochlore structure has been studied by several researchers. It is reported that the phonon modes corresponding to the O′–A–O′and O–A–O bending modes account for almost 80%of the total dielectric permittivity.

Fig.2. Raman spectra of BMN and calculated Raman-active mode intensity.
X-ray photoelectron spectroscopy was also applied to reveal the chemical states of all elements in BMN pyrochlores.The XPS spectrum of oxygen to 1s orbital is shown in Fig.3(e),the total peak can be present as a sum of two gaussian peaks using Fourier transform,which suggests that there are two kinds of O2−anions in different chemical environment in BMN pyrochlores. The ratio of the two gaussian peak areas is 1:5.998,it agreed perfectly with the ratio of two kinds of O atoms in the center of A2O′tetrahedrons and vertex of BO6octahedrons. DFT results suggest a higher charge transfer were observed between B cations and O anions in B–O bonds than cations and anions in A–O′bonds. Therefore,the binding energy of O in B–O bonds 1s spectrum(528.9 eV)is lower than its counterpart (532.5 eV) in A–O′bonds. The Bi 4f orbital in high-resolution XPS spectrum is shown in Fig.3(b). the total intensity of XPS spectrum can be split into two gaussian peaks, these two peaks attribute to the two orbitals 4f7/2and 4f5/2in 4f energy level, the higher peak in 158.1 eV is assigned to the 4f7/2orbital and another peak in 162.1 eV is assigned to 4f5/2orbital.It is the evidence indicating the bismuth in BMN pyrochlores were tri-valent. Also, the Nb 3d XPS spectrum (Fig. 3(d)) also have two peaks assigned to 3d3/2(208.5 eV)and 3d5/2(205.7 eV),indicating the Nb cations are penta-valent. No impure phase was found in the result. The overall XPS analysis of all elements result suggests that the ceramic sample is well crystallized and the cubic pyrochlore is the main phase in the BMN ceramics.
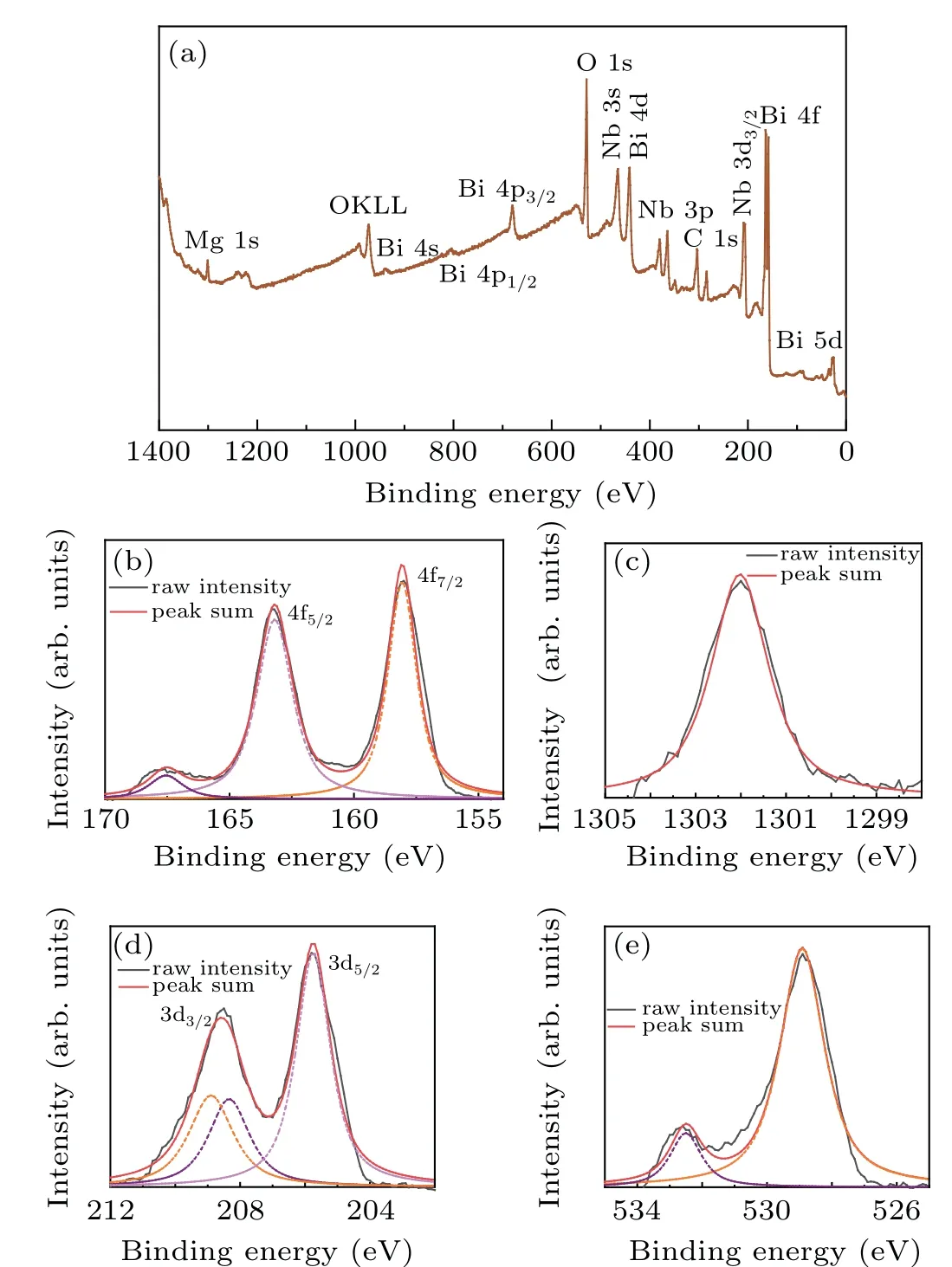
Fig. 3. (a) XPS spectra and all elements XPS spectrum in BMN, (b) Bi 4f orbital XPS spectrum,(c)Mg 1s orbital XPS spectrum,(d)Nb 3d orbital XPS spectrum,and(e)O 1s orbital XPS spectrum.
Berry Phase calculations give out how the A2O′tetrahedron structure distorts before and after the application of the external electric field. The main axe of the BO6octahedra is not perpendicular to any of the basic axis of the whole crystal structure as mentioned before,so does the A2O′structure. Experimentally, it is known that the bismuth ions displace from their ideal crystallographic site for some simple bismuth pyrochlores. Due to the symmetry of our initial structure, the force examination after the calculations means that only the O-48f atoms have non-zero forces at the initial structure. The polarization of BMN was calculated in Berry phase calculations,BMN under the zero bias electric field has the same polarization value in three directions. Polarization on thec-axis soars to 10707.851 as a consequence of applying an external field on thec-axis direction.The theoretical dielectric constant is described using the formula below:[28]

whereDis the total dipole,D(0)is the dipole under zero bias,Eis the applied electric field intensity, andΩis the volume of the cell. The theorical dielectric constant of BMN material is 132.78. Considering other mechanisms such as atomic hopping was neglected in calculations,this result is acceptable.
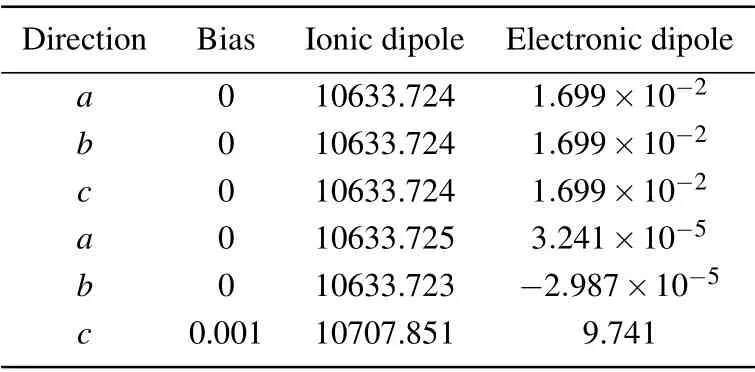
Table 2. Polarization on each direction.
The Bader charge of BMN before and after applying electric field are also investigated. All the atoms in BMN was set to relax while the lattice constant was fixed. After relaxation calculation,all cations and anions are exhibiting displacement from its initial position as well as charge transfer under 0.001 Ry a.u. electric field alongc-axis. An obvious difference in atomic displacements between A-site cations and B-site cations can be observed in Fig. 5(a). The A-site Mg cations have the highest displacement(0.765028 °A),followed by Bi cations(0.346317 °A).The A-site(Bi/Mg)displacements are higher than their counterparts of B-site(Nb/Mg). This result proves that the A2O′tetrahedrons are more flexible than the BO6octahedrons under the external field.
Charge transfer can somehow relieve the Coulomb force exerted on both cations and anions, which can be proved by comparison of Nb and B-site Mg. Nb atoms have the highest average charge transfer (0.02508e) among all the cations,but possess a medium displacement in all species. In conclusion, the A-site cations incline to change atomic positions in response to the external field, the A-site cations have higher ionic polarization than the B-site cations. BO6octahedra are more stable than the A2O′tetrahedrons under the external field. Comparing to Nb cations, B-site Mg cations have a lower displacement and charge transfer,which refers to a more stable structure. The Mg dopant in the A-site can result in a higher displacement in the electric field and more obvious distortion to the A2O′structure, thus the A–O′and A–O bonds are more sensitive to the external field with Mg cations introduced into A-sites.
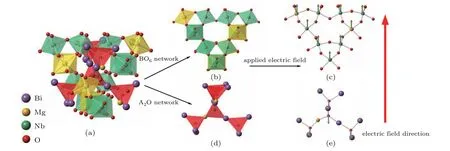
Fig.4. (a)BO6 network and A2O′ network in BMN,(b)BO6 network, (c)distortion in BO6 network after applying electric field, (d)A2O′ network,and(e)distortion in A2O′ network after applying electric field.

Fig.5. (a)Cation displacement and charge transfer,(b)dielectric permittivity and the loss of BMN thin films.
Due to the sensitive response of A–O and A–O′bonds to the external feilds and the signifciant contribution to dielectric permittivity,the dielectric permittivity of BMN thin flims decreased sharply when the external electric feild intensity went up. In this study,the tunability in dielectric constant of BMN ceramic was measured and present by the thin flim sample deposited on the sapphire substrate at a temperature of 700°C.The total sputter gas pressure of 1 Pa with O2/(O2+Ar)mixing ratio of 15% was maintained during the whole sputtering procedure. The tunability of BMN thin films was measured by an impedance analyzer with two probes closely contacting silver electrodes on the surface of thin films. A bias field was applied on the two electrodes and gradually increased. Capacitance and dielectric loss were measured until the bias field reaches the breaking point.Figure 5(b)depicts the relationship between the bias electric field and dielectric constant,and the dielectric constant drops as the applied bias electric field goes up and maximum tunability of 43%is achieved at an applied bias field of 1.5 MV/cm. The loss tangent is lower than 0.008 in a statistical average. It slightly grows when the bias field is over 1.0 MV/cm, which can be attributed to the leakage loss caused by the high bias field.
4. Conclusion
The cubic pyrochlore-structure BMN ceramics were synthetized by a conventional solid-state reaction,and BMN thin films were prepared on sapphire substrates by rf magnetron sputter deposition. The BMN thin film’s dielectric property and tunability were investigated.The maximum dielectric tunability (~43%) of BMN thin films was achieved at a bias field of 1.5 MV/cm. Berry Phase calculation results depict the atomic displacement under the applied electric field and give out the theoretical dielectric constant contributed by displacement. The A2O′tetrahedrons are more easy to distort under external field. The A-site Mg has the highest displacement(0.765028 °A),followed by Bi cations(0.346317 °A),which is also in the A-site,this distortion greatly infects the A–O′and A–O bonds in length and covalency. Coulomb force on A-site cations also places a damping factor on bond vibration,which explains the high tunability of BMN materials.
猜你喜欢
杂志排行

Chinese Physics B的其它文章
- Physical properties of relativistic electron beam during long-range propagation in space plasma environment∗
- Heterogeneous traffic flow modeling with drivers’timid and aggressive characteristics∗
- Optimized monogamy and polygamy inequalities for multipartite qubit entanglement∗
- CO2 emission control in new CM car-following model with feedback control of the optimal estimation of velocity difference under V2X environment∗
- Non-peripherally octaalkyl-substituted nickel phthalocyanines used as non-dopant hole transport materials in perovskite solar cells∗
- Dual mechanisms of Bcl-2 regulation in IP3-receptor-mediated Ca2+release: A computational study∗