敲低PLK1对脉络膜黑色素瘤细胞增殖的抑制作用及其机制
2021-10-21赵芃芃白小芳卢凤丽李思园蒋胜群
赵芃芃,白小芳,卢凤丽,李思园,谭 丛,蒋胜群,郁 佳,秦 梅
(蚌埠医学院第一附属医院眼科,蚌埠 233000;*通讯作者,E-mail:bbmcqm@163.com)
脉络膜黑色素瘤(chroidal melanoma,CM)是成人眼内最常见的恶性肿瘤,在全球发病率占眼内恶性肿瘤的首位,恶性程度及致死率均较高,中位生存时间仅为3-5月[1]。目前,CM最主要的两种治疗方法有两种:手术和放射性敷贴治疗。发病早期可造成视力损害,晚期则眼外蔓甚至危及生命[2],但由于发病隐匿,发现时多为晚期,故而早期诊断治疗显得尤为重要。PLK1属于polo样激酶家族中的一员,大量研究表明PLK1在人类肿瘤发生发展中具有重要作用,但PLK1在脉络膜黑色素瘤中的作用国内外尚无研究报道。因此,本研究拟通过分子生物学和表观遗传学方法探究PLK1对CM细胞增殖的影响及其可能分子机制,以期为CM的临床诊断和治疗提供新的靶点及思路。
1 材料和方法
1.1 主要试剂
RPMI-1640培养基、胎牛血清、胰蛋白酶消化液购于Gibco生物公司(美国);CCK-8试剂盒、青链霉素双抗溶液、二甲基亚砜(dimethyl sulfoxide,DMSO)溶液、凝聚胺溶液购自睿捷生物(合肥);PLK1抗体购自Abcam公司(美国);BUBR1、GAPDH抗体购于武汉三鹰生物技术公司;组蛋白H3、H3T3ph抗体购于杭州景杰生物公司;RT-qPCR试剂盒购于南京诺唯赞生物公司;细胞周期检测试剂盒购于南京凯基生物公司;PLK1敲低慢病毒购于上海吉玛基因生物公司;染色质免疫共沉淀(chromatin immunoprecipitation,ChIP)检测试剂盒购于碧云天生物公司(上海);所有引物由滁州通用生物公司合成。
1.2 细胞培养
人脉络膜黑色素瘤C918细胞购于南京凯基生物公司,人脉络膜血管内皮细胞、人脉络膜黑色素瘤MuM-2B、MuM-2C细胞购于武汉普诺赛生命科技有限公司。脉络膜黑色素瘤细胞复苏后转移至含有10%胎牛血清和1%青链霉素双抗溶液的RPMI-1640培养基;脉络膜血管内皮细胞复苏后转移至普诺赛血管内皮细胞专用培养基,置于37 ℃、5% CO2细胞孵箱中常规培养。
1.3 PLK1 shRNA慢病毒感染细胞
为构建PLK1敲低细胞模型,选取对数生长期的C918细胞,消化计数。接种于6孔板中,调整细胞浓度使每孔细胞数量为4×105,细胞培养箱继续培养24 h进行病毒感染,实验分为对照组、PLK1敲低组1(sh1组)、PLK1敲低组2(sh2组),即分别加入阴性对照慢病毒(NC组)、PLK1 shRNA1慢病毒、PLK1 shRNA2慢病毒,每孔加入20 μl不同的慢病毒原液和1‰凝聚胺溶液,继续培养24 h更换新鲜培养基、48 h收集细胞用于后续实验。
1.4 CCK-8法检测细胞增殖
为检测敲低PLK1对C918细胞增殖的影响,按1.3分组及处理,选取慢病毒感染后生长状态良好的C918细胞,消化计数、调整细胞密度,将其接种至96孔板中,每孔加入100 μl含有2 000个细胞的培养液,每组设置6个复孔。在细胞培养箱中继续分别培养0,24,48,72 h,每孔加入10 μl CCK-8溶液,培养箱中继续培养1.5 h,酶标仪检测各组吸光度OD450值。
1.5 克隆形成实验检测细胞增殖
为检测细胞克隆形成能力,按1.3分组及处理,选取感染后生长状态良好的C918细胞消化计数、调整细胞密度,将其接种至6孔板中,每孔1 000个细胞,每组设置3个复孔。在细胞培养箱中继续培养10 d弃去培养基,甲醇室温固定20 min,结晶紫染色30 min,蒸馏水洗净烘干,拍照记录结果。
1.6 细胞周期检测
为检测PLK1敲低后对细胞周期的影响,按1.3分组及处理,选取感染后的C918细胞,消化收集细胞,用PBS洗涤一次,调整细胞密度为1×106/ml,取1 ml单细胞悬液,2 000 r/min离心5 min,弃去上清。在细胞中加入体积分数为70%的预冷乙醇500 μl,4 ℃过夜固定。染色前用预冷PBS洗去固定液,加入提前配置好的500 μl染色工作液(临用前将核糖核酸酶A和碘化丙锭按1 ∶9配制),室温避光染色50 min,流式细胞仪检测激发波长488 nm处红色荧光。所得结果使用Flowjo 7.6软件进行细胞周期分析。
1.7 RT-qPCR检测PLK1、BUBR1、DDX11、DDX21、TFDP2、CHEK1、CHEK2基因mRNA表达水平
将按1.3分组及处理后的C918细胞收集于1.5 ml无酶EP管中,加入1 ml Trizol提取RNA,逆转录成cDNA。参照南京诺唯赞RT-qPCR试剂盒配制反应体系,上机检测。反应条件为:95 ℃ 5 min,95 ℃ 10 s,60 ℃ 30 s,反应40个循环。引物序列见表1。产物与内参比较,采用2-ΔΔCt相对定量的方式对结果进行计算。

表1 RT-qPCR实验所使用的引物序列
1.8 Western blot分析PLK1、BUBR1蛋白的表达
将细胞接种于60 mm平皿中(2×105个/孔),培养18-24 h后,按1.3分组及处理,消化收集细胞,用预冷PBS洗2次,2 000 r/min离心10 min后弃去上清液,根据细胞量加入预冷的蛋白裂解液,冰上裂解30 min。4 ℃低温离心机中12 000g离心30 min提取细胞总蛋白,BCA蛋白定量法测定各组蛋白浓度,调整蛋白浓度,煮样变性。每组加入30 μg蛋白,SDS-PAGE电泳(70 V 30 min,120 V 70 min);转膜至PVDF膜;5%脱脂牛奶室温封闭1 h;一抗(PLK1抗体按1 ∶1 000稀释、BUBR1抗体按1 ∶2 000稀释、GAPDH抗体按1 ∶50 000稀释)4 ℃孵育过夜;PBST洗膜3次,30 min/次;二抗室温孵育1 h;PBST洗膜3次,30 min/次;ECL发光试剂盒显影;Bio-Rad凝胶成像系统获取图像,收集数据。
1.9 组蛋白提取
为检测PLK1敲低后对组蛋白H3第3位苏氨酸磷酸化(H3T11ph)的影响,按1.3分组及处理,收集感染后状态良好的C918细胞,使用预冷的PBS洗涤2次,弃去上清,加入适量体积的聚乙二醇辛基苯基醚提取缓冲液(triton extraction buffer,TEB)溶液重悬细胞,冰上裂解10 min,6 500g,4℃离心10 min收集细胞核,弃去上清。再用TEB溶液洗涤细胞核,离心,弃去上清。用0.2 mol/L盐酸重悬细胞核,4 ℃过夜提取。6 500g,4 ℃离心10 min,上清为提取的组蛋白,-20 ℃保存、备用。
1.10 ChIP实验检测组蛋白H3T3ph修饰在下游靶基因启动子上的富集情况
为检测H3T3ph修饰在靶基因启动子区域的富集情况,进行ChIP实验。按照碧云天生物公司ChIP检测试剂盒说明书进行具体操作,简述如下。选取细胞生长状态较好的C918细胞,收集于15 ml离心管中,使用10 ml无血清培养基重悬细胞,加入终浓度1%的甲醛,室温交联固定DNA与蛋白复合体10 min,随后加入终浓度为0.12 mol/L的甘氨酸终止交联,冰上制备细胞核,然后重悬细胞核并将其转入新的EP管进行超声剪切染色质,14 000 r/min,4 ℃离心10 min收集上清,取5 μl上清液作为Input对照组,放入-80 ℃保存备用。另外各取45 μl上清分别加入2 μg IgG抗体(IgG组)和组蛋白H3T3ph修饰抗体(H3T3ph组),且均加入protein G珠子4 ℃摇床过夜。4 ℃离心收集沉淀并洗涤,随后洗脱免疫沉淀复合物,每个样加入2 μl蛋白酶K和16 μl的5 mol/L NaCl溶液,65 ℃过夜消化解交联。最后使用酚氯仿提取DNA用于后续PCR定量实验。通过比较H3T3ph组的信号强度相比于IgG组信号强度在靶基因启动子上的富集倍数,进而明确H3T3ph修饰在靶基因启动子上的结合位置。
1.11 统计学方法
实验数据均使用SPSS 20.0软件进行处理分析,数据均以平均数±标准差表示,多组间差异采用单因素方差分析,两组间差异比较使用t检验。P<0.05认为差异具有统计学意义。
2 结果
2.1 PLK1在脉络膜黑色素瘤细胞中高表达
RT-qPCR结果表明:相比于脉络膜血管内皮细胞,脉络膜黑色素瘤MuM-2B、MuM-2C、C918细胞的PLK1 mRNA表达显著升高,且差异具有统计学意义(P<0.05,见图1A)。Western blot结果证实:相比于脉络膜内皮细胞,脉络膜黑色素瘤细胞中PLK1蛋白表达水平上调(见图1B)。因PLK1在C918细胞中表达量最高,故将C918细胞用于后续实验。

图1 PLK1在脉络膜血管内皮细胞和脉络膜黑色素瘤细胞中表达情况Figure 1 The expression of PLK1 in choroidal vascular endothelial cells and choroidal melanoma cells
2.2 构建PLK1敲低的脉络膜黑色素瘤C918细胞模型
RT-qPCR实验表明:相比于Scr对照组,sh1、sh2组的PLK1 mRNA水平显著减少,且差异具有统计学意义(P<0.05,见图2A)。同时,我们也进行了Western blot实验,结果表明:与NC组比较,sh1、sh2组的PLK1蛋白水平明显降低(见图2B)。RT-qPCR实验和Western blot实验结果证实C918细胞的PLK1敲低模型构建成功。

图2 C918细胞感染PLK1慢病毒后敲低效果验证Figure 2 Validation of knockdown efficacy in C918 cells infected with PLK1 lentivirus
2.3 敲低PLK1抑制C918细胞增殖
CCK-8实验实验结果显示:随着时间的延长,相比于NC组,sh1组和sh2组的细胞增殖能力逐渐减弱,且在72 h时差异具有统计学意义(P<0.05,见图3A)。随后的克隆形成实验结果也表明:相比于NC组,sh1组和sh2组的克隆数目明显减少(P<0.05,见图3B)。
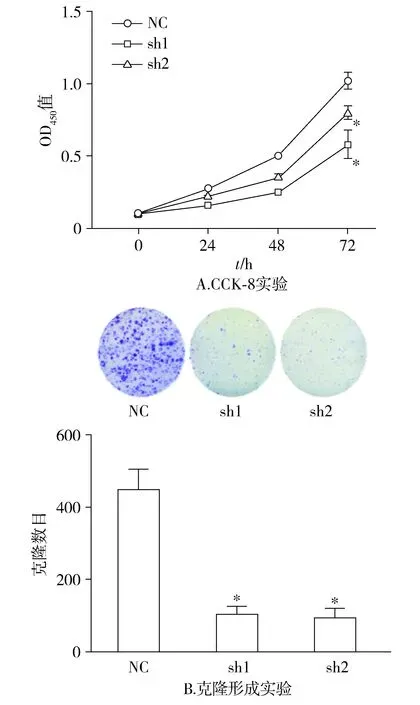
与NC组比较,*P<0.05图3 敲低PLK1对C918细胞增殖的影响Figure 3 The effect of knocking down PLK1 on the proliferation of C918 cells
2.4 敲低PLK1后将细胞周期阻滞于S期
流式细胞仪检测结果证实:相比于NC组,sh1组和sh2组处于S期的细胞比例明显增多,G2/M细胞比例明显减少,且差异具有统计学意义(P<0.05,见图4)。

图4 敲低PLK1对C918细胞周期的影响Figure 4 The effect of PLK1 knockdown on the cell cycle of C918
2.5 敲低PLK1降低BUBR1基因的转录
RT-qPCR结果表明PLK1敲低后BUBR1 mRNA水平显著下降(P<0.05,见图5)。
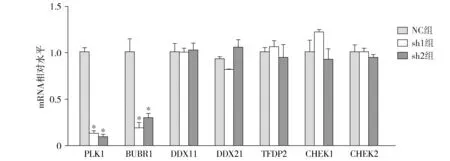
与NC组比较,*P<0.05图5 RT-qPCR检测敲低PLK1表达后其可能的下游靶基因变化Figure 5 Expression of possible downstream target genes after knocking down PLK1 by RT-qPCR
2.6 敲低PLK1减少BUBR1的蛋白表达水平
Western blot实验检测BUBR1的蛋白表达,结果显示:相比于NC组,sh1组和sh2组的PLK1蛋白表达量明显减少(见图6)。
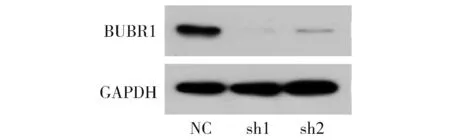
图6 敲低PLK1对BUBR1蛋白表达的影响Figure 6 The effect of knocking down PLK1 on the expression of BUBR1 protein
2.7 敲低PLK1减少组蛋白H3T3ph修饰水平
Western blot结果证实:敲低PLK1后H3T3ph修饰的表达水平明显降低(见图7)。

图7 敲低PLK1对组蛋白H3T3ph修饰的影响Figure 7 The effect of knockdown of PLK1 on histone H3T3ph modification
2.8 组蛋白H3T3ph修饰能够富集在BUBR1基因的启动子上
ChIP实验结果表明:相比于IgG,组蛋白H3T3ph修饰能够富集在引物3处,即启动子上游-500 bp至-1 000 bp区域(P<0.05,见图8)。
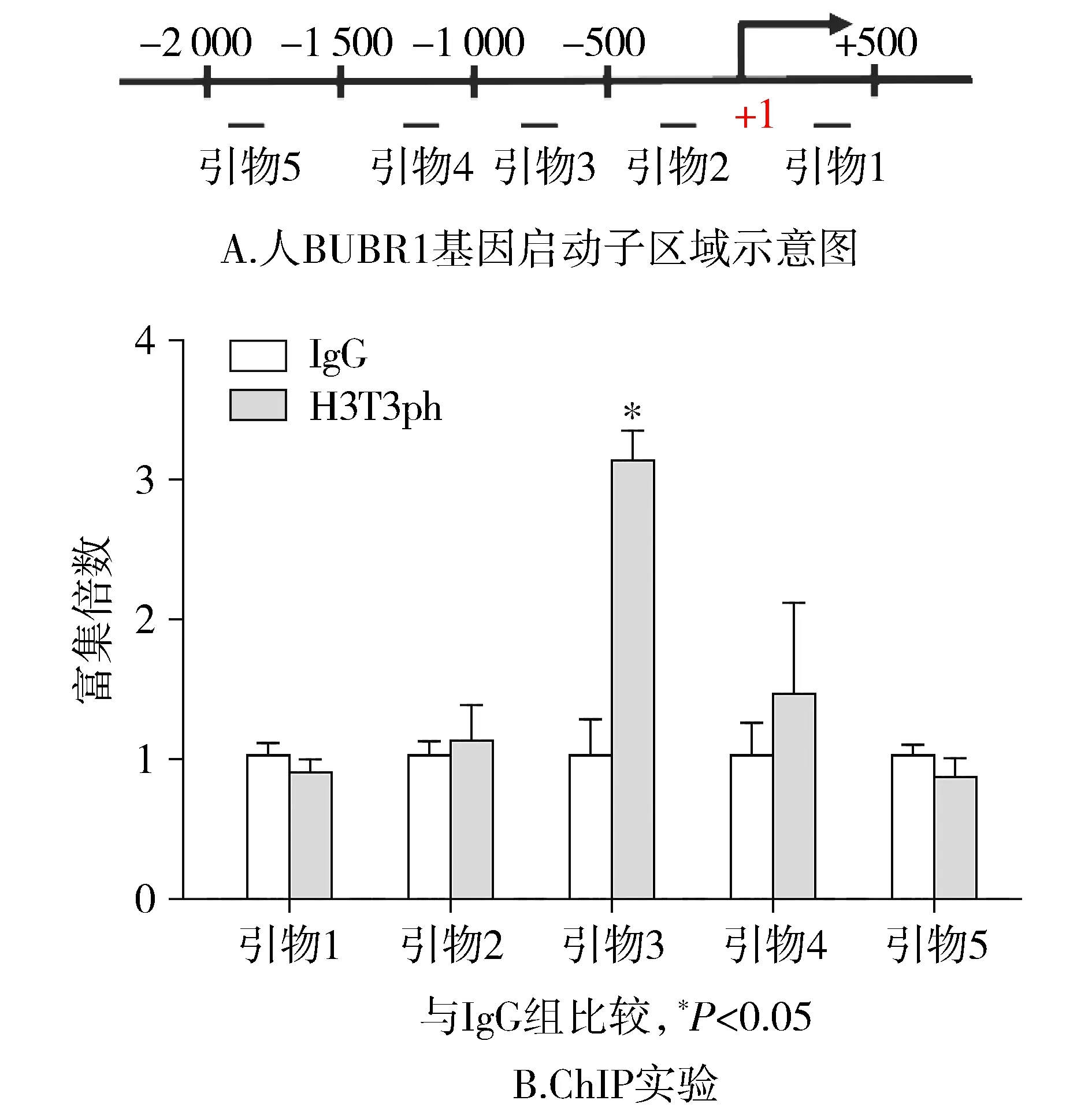
图8 组蛋白H3T3ph修饰在BUBR1基因的启动子上的富集情况Figure 8 Enrichment of histone H3T3ph modification on the promoter of BUBR1 gene
3 讨论
polo样激酶家族(polo-like kinases,PLKs)是一类结构和功能均高度保守的丝/苏氨酸蛋白激酶,可参与细胞周期不同阶段的调控。哺乳动物中PLKs分为5个亚型,结构具有相似性,即N末端为高度保守的激酶结构域,其是一个催化T-loop,允许PLKs将ATP转化为ADP并将磷酸基转移到PLKs下游的任何一个磷酸化目标上[3];C端为催化活性及亚细胞动态定位的特征性PBD(polo-box domain)结构域,能够特异性识别磷酸化位点,是PLKs定位与靶向的关键[4]。PLK1是polo样激酶家族中备受关注的成员,能够调控细胞周期,并能促进中心体成熟、染色体分裂和胞质分裂等,其功能的实现可能与下游蛋白(FoxM1、Myc、Mdm2、Pten及P53等)的相互作用和激活有关[5]。目前研究发现,PLK1在多种肿瘤中高表达,包括肺癌、头颈部肿瘤、食管癌、胃癌、乳腺癌及前列腺癌[6];并且有研究表明PLK1能够参与顺铂耐药[7],提示PLK1在人类肿瘤发生发展中具有重要作用,但关于PLK1在脉络膜黑色素瘤中的作用尚未见相关研究报道。
我们首先证实PLK1在脉络膜黑色素瘤细胞中高表达,且在C918细胞中表达量最高;随后构建脉络膜黑色素瘤的PLK1敲低的C918细胞模型,RT-qPCR和Western blot实验显示PLK1 shRNA慢病毒敲低效果较好,证实PLK1敲低细胞模型构建成功。随后通过CCK-8和克隆形成实验证实,敲低PLK1能显著抑制C918细胞增殖,且能将细胞周期阻滞于S期,使得细胞无法正常向G2/M期分裂。为探究PLK1调控脉络膜黑色素瘤细胞增殖的分子机制,我们使用RT-qPCR检测了多种细胞增殖相关基因的变化,结果表明敲低PLK1可以减少BUBR1的mRNA水平,Western blot实验结果显示敲低PLK1能显著下调BUBR1的蛋白表达水平。
BUBR1作为有丝分裂检查点中一个极其关键的蛋白,在染色体的分离、排列和细胞分裂过程中发挥重要功能,从而保证细胞周期的顺利进行[8]。已有研究证实,BUBR1在食管癌、肾癌、卵巢癌、胃癌、膀胱癌,乳腺癌中异常高表达[9-14],且与肿瘤病人的不良预后具有相关性。我们的研究初步证实PLK1可能是通过调控BUBR1基因的表达进而促进络膜黑色素瘤细胞的增殖。紧接着我们进一步探究PLK1调控BUBR1基因表达的分子机制。
组蛋白磷酸化(histone phosphorylation)指在组蛋白的N末端结合带有负电荷的磷酸基团,是真核生物最广泛的蛋白修饰之一。这一过程是可逆的,磷酸化修饰常发生在组蛋白的丝氨酸/苏氨酸残基上。许多组蛋白的丝氨酸或苏氨酸残基磷酸化与基因转录激活、细胞周期依赖的染色体缩合、DNA修复和凋亡诱导的染色质致密化有关[15]。组蛋白磷酸化修饰主要由Haspin、MSK1、MSK2、CKII、Mst1等核蛋白激酶直接催化介导。已有研究证实PLK1能够调控Haspin进而激活组蛋白H3第三位苏氨酸的磷酸化(H3T3ph)修饰[16],提示PLK1可能在基因转录调控中发挥重要功能。Western blot结果表明敲低PLK1可以减少C918细胞中的组蛋白H3T3ph修饰表达水平,并且ChIP实验证实H3T3ph能够富集在BUBR1的启动子上游-500 bp至-1 000 bp区域,这些结果说明PLK1很可能是通过介导H3T3ph修饰在BUBR1的启动子上富集来调控BUBR1的基因转录,但这一结论仍需通过更多实验进行验证。
综上,本研究初步证实PLK1能促进脉络膜黑色素瘤细胞的增殖,其机制可能与PLK1调控BUBR1基因的转录相关。因此,本研究为PLK1用于脉络膜黑色素瘤的早期诊断和治疗提供新的思路和参考。
