Hepatocellular carcinoma in nonalcoholic fatty liver disease: A growing challenge
2021-10-11ngeloMattosJoseDebesRenuDhanasekaranJihaneBenhammouMarcoArreseAndrLuizPatrcioAmandaZilioAngeloMattos
Ângelo Z Mattos, Jose D Debes, Renu Dhanasekaran, Jihane N Benhammou, Marco Arrese, André Luiz V Patrício, Amanda C Zilio, Angelo A Mattos
Ângelo Z Mattos, Angelo A Mattos, Graduate Program in Medicine: Hepatology, Federal University of Health Sciences of Porto Alegre, Porto Alegre 90020-090, Rio Grande do Sul, Brazil
Ângelo Z Mattos, André Luiz V Patrício, Amanda C Zilio, Angelo A Mattos, Gastroenterology and Hepatology Unit, Irmandade Santa Casa de Misericórdia de Porto Alegre, Porto Alegre 90020-090, Rio Grande do Sul, Brazil
Jose D Debes, Department of Medicine, Division of Infectious Diseases and of Gastroenterology, University of Minnesota, Minneapolis, MN 55455, United States
Jose D Debes, Department of Gastroenterology and Hepatology, Erasmus Medical Center, Rotterdam 3015 CN, South Holland, Netherlands
Renu Dhanasekaran, Division of Gastroenterology and Hepatology, Stanford University, Stanford, CA 94305, United States
Jihane N Benhammou, The Vatche and Tamar Manoukian Division of Digestive Diseases, University of California, Los Angeles, CA 90095, United States
Marco Arrese, Department of Gastroenterology, Pontificia Universidad Católica de Chile, Santiago 3580000, Chile
Abstract Nonalcoholic fatty liver disease (NAFLD) is the most common cause of liver disease worldwide, and its prevalence increases continuously. As it predisposes to hepatocellular carcinoma both in the presence and in the absence of cirrhosis, it is not surprising that the incidence of NAFLD-related hepatocellular carcinoma would also rise. Some of the mechanisms involved in hepatocarcinogenesis are particular to individuals with fatty liver, and they help explain why liver cancer develops even in patients without cirrhosis. Genetic and immune-mediated mechanisms seem to play an important role in the development of hepatocellular carcinoma in this population. Currently, it is consensual that patients with NAFLD-related cirrhosis should be surveilled with ultrasonography every 6 mo (with or without alpha-fetoprotein), but it is known that they are less likely to follow this recommendation than individuals with other kinds of liver disease. Moreover, the performance of the methods of surveillance are lower in NAFLD than they are in other liver diseases. Furthermore, it is not clear which subgroups of patients without cirrhosis should undergo surveillance. Understanding the mechanisms of hepatocarcinogenesis in NAFLD could hopefully lead to the identification of biomarkers to be used in the surveillance for liver cancer in these individuals. By improving surveillance, tumors could be detected in earlier stages, amenable to curative treatments.
Key Words: Nonalcoholic fatty liver disease; Nonalcoholic steatohepatitis; Hepatocellular carcinoma; Hepatocarcinogenesis; Surveillance
INTRODUCTION
Nonalcoholic fatty liver disease (NAFLD) is rapidly becoming one of the most common causes of liver disease worldwide[1]. According to a meta-analytic assessment of 86 studies, the global prevalence of NAFLD is 25.24%[2]. Therefore, its association with hepatocellular carcinoma (HCC) also becomes increasingly important[3]. The relevance of this association is demonstrated by the fact that NAFLD was responsible for 36300 incident cases of HCC and 34700 HCC-related deaths in 2019[4].
Although cirrhosis is considered a predisposing condition for HCC in general, diverse disease-specific mechanisms are involved in the development of NAFLDrelated HCC[3,5,6]. Moreover, the observation that HCC can occur in patients with NAFLD even in the absence of cirrhosis suggests that, as in the case of hepatitis B virus infection, NAFLD itself could be etiologically linked to HCC development[7]. Over the last few years, an array of studies has shed light on the diverse genetic and immunerelated mechanisms that link NAFLD to the process of hepatocarcinogenesis. Nonetheless, much work is still needed to further understand this inter-relation.
Considering the association between NAFLD and HCC, surveillance for liver cancer among patients with fatty liver has become an important topic of discussion. However, the extremely high prevalence of NAFLD and the distinct risk levels for HCC in different patients make defining the target population for surveillance quite challenging[8].
The aim of this article is to review the epidemiology of NAFLD-related HCC, the genetic and immune mechanisms involved in hepatocarcinogenesis in individuals with NAFLD, the current knowledge related to HCC in patients with NAFLD without cirrhosis, and key aspects to consider for HCC surveillance in NAFLD.
EPIDEMIOLOGY OF NAFLD-RELATED HCC
In the last few decades, HCC-related mortality has steadily increased and since the 1980s has almost tripled in the United States, where it is the fastest-rising cause of cancer-related death[9]. Notably, this increase parallels the growth in NAFLD prevalence, which increased 2 to 3-fold in a similar period of time[10], turning it into a leading etiology of cirrhosis worldwide[11]. These coinciding trends and the fact that NAFLD has been noted as an increasingly common cause of HCC in several series[12] as well as the fastest-growing cause of HCC in liver transplant candidates and recipients in the United States[13] suggest that NAFLD is a prominent contributor to HCC burden worldwide and that the prevalence of HCC will likely increase concomitantly with the global obesity epidemic[12,14]. In this context, a recent study used Bayesian models to estimate that the age-standardized incidence rate of NAFLDrelated liver cancer would increase from 0.92/100000 inhabitants in 2018 to 1.18/100000 inhabitants in 2030[15].
Estimates regarding the annual incidence of HCC in patients with NAFLD-related cirrhosis in the western hemisphere range from 0.5% to 2.6%[14,16]. With regard to data from eastern hemisphere countries, a prospective study from Japan reported similar figures, with an annual incidence of 2.26% in a cohort followed for more than 15 years[17]. Another study from India reported lower figures (annual incidence of HCC of 0.5% in patients with biopsy-proven NAFLD-related cirrhosis)[18]. It is worth mentioning, though, that most of these estimates originate from cohorts followed in tertiary centers or from liver transplant registries and that population-based cohort studies are not available. Importantly, existing data suggest that older age, male sex, alcohol intake, and especially diabetes are factors that may increase HCC incidence in NAFLD-related cirrhosis[19]. The annual incidence of HCC among individuals with NAFLD who do not have cirrhosis is much lower than that reported for patients with cirrhosis, as it will be reviewed later in this article.
GENETIC ASPECTS OF NAFLD-RELATED HCC
Considering the particular characteristics of NAFLD and NAFLD-related HCC as well as the fact that liver cancer also develops in individuals with NAFLD who do not have cirrhosis, the study of the genetic aspects of hepatocarcinogenesis in NAFLD has drawn substantial attention. The main genetic mechanisms involved in the development of NAFLD-related HCC will be discussed in this section and are summarized in Figure 1.
Genetic variants associated with NAFLD-related HCC
Early NAFLD studies have identified ethnic differences and evidence of familial clustering suggestive of a hereditary/genetic component to the disease[20]. The first study to demonstrate an association between genetic variants and NAFLD was published by Romeoet al[21] who conducted a genome wide association analysis using quantitative proton magnetic resonance spectroscopy to measure hepatic steatosis. The genome wide association analysis showed that carriers of the rs738409 variant of the patatin-like phospholipase domain containing protein 3 (PNPLA3) gene, most commonly found among Hispanics, had over a 2-fold increase in intrahepatic triglycerides[21]. Subsequent studies demonstrated the same variant to be associated with NAFLD-related HCC[22,23].
Following studies described conflicting evidence of an association between the transmembrane 6 superfamily member 2 (TM6SF2) rs58542926 polymorphism and NAFLD-related HCC, potentially from its low minor allele frequency[24,25]. The membrane bound O-acetyltransferase domain containing 7 rs641738 variant was posteriorly identified in a European cohort to be associated with NAFLD-related HCC[25-30]. Another European study focusing on the identification of rare variants in NAFLD-related HCC cases found, aside from PNPLA3 and TM6SF2, pathogenic variants in apolipoprotein B gene, among others[31]. As genetic association studies have mostly included patients of European ancestry, larger and more diverse cohorts are needed given the clinical observation that Hispanics are at higher risk for NAFLDrelated HCC[32].
Molecular events in NAFLD-related hepatocarcinogenesis
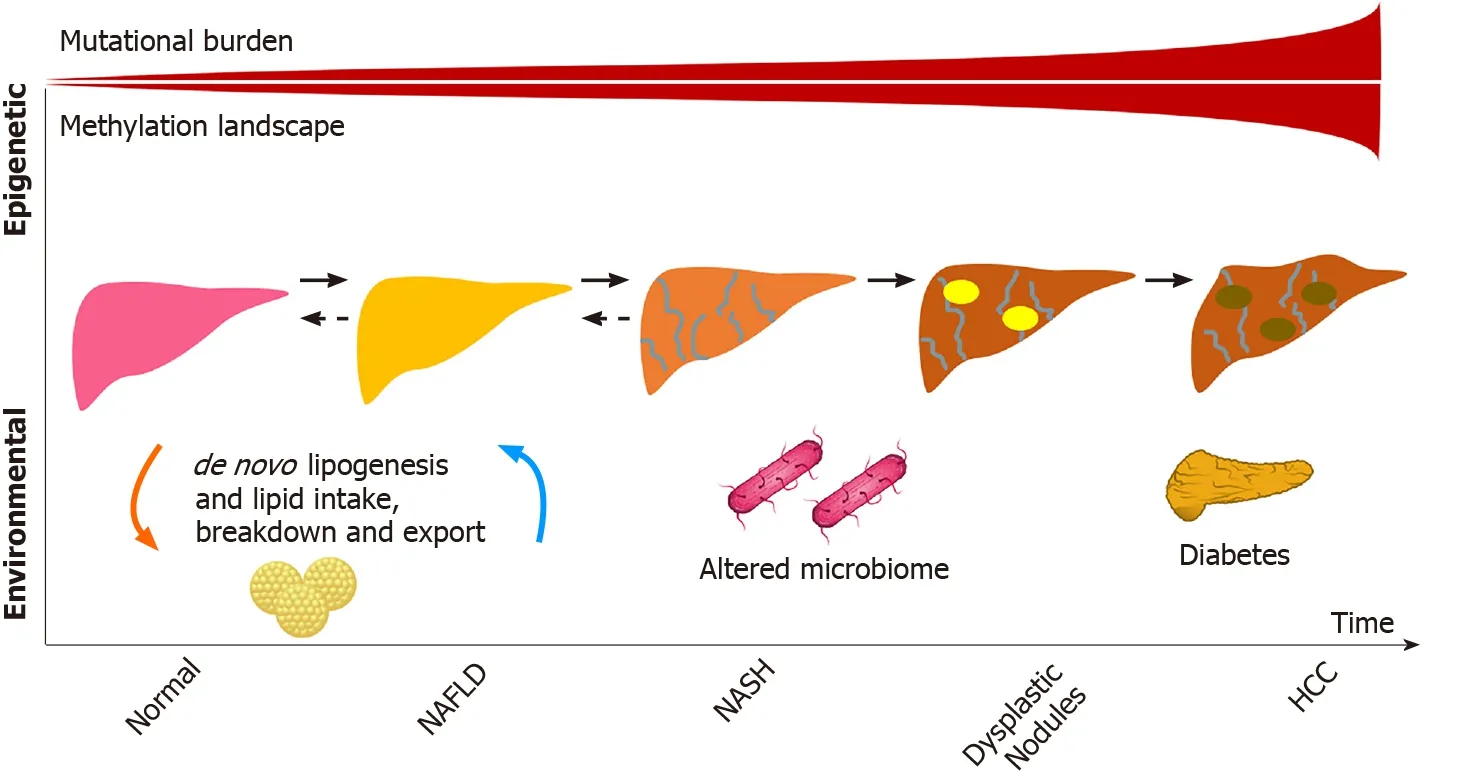
Figure 1 Main genetic factors determining nonalcoholic fatty liver disease-related hepatocarcinogenesis
Association studies have provided a plethora of information regarding NAFLDrelated hepatocarcinogenesis, although mechanistic studies have yet to elucidate how these variants cause disease. The observation that many of the polymorphisms involve lipid regulation raises the possibility that a lipid-rich dysregulated microenvironment may be key to HCC development. Although NAFLD-specific HCC studies are lacking, parallel mutations exist between NAFLD and other etiologies demonstrating a potential convergence in pathways that have previously been described in viral etiologies[33]. For instance, mutations in telomerase reverse transcriptase are known to play a role in the progression of dysplastic nodules and in the development of early HCC[34,35].
As hepatocyte damage increases from cirrhosis to dysplasia and eventually HCC, the mutational burden leading to cancer exponentially grows. This was well illustrated in a study by Brunneret al[36] who conducted whole genome sequencing of 100-500 hepatocytes from 5 healthy controls and 9 patients with cirrhosis. Structural variants and copy number variations were more commonly identified in those with cirrhosis compared to the normal controls, including in activin receptor type 2A, cyclindependent kinase inhibitor 2A, and AT-rich interaction domain 5A. Interestingly, similar signatures of somatic copy number variations were identified in a pilot study of 10 HCC cases in circulating tumor cells, raising the possibility of their use as biomarkers[37]. Other well described pathways include mutations in β-catenin, tumor antigen p53, and AKT/mechanistic target of rapamycin/mitogen-activated protein kinase signaling pathway, which includes tuberous sclerosis complex subunits 1 and 2, phosphatase and tensin homolog, and fibroblast growth factor 19[34].
Given the clinical and genetic heterogeneity in human HCCs, animal models have provided the pre-clinical tools to understand these pathways in NAFLD-related HCC[38]. Although NAFLD and nonalcoholic steatohepatitis (NASH) mouse models have limitations in recapitulating the human NAFLD phenotype, these animal models have proven especially relevant when comparing “obese” and “lean” NAFLD-related HCCs. Using whole exome sequencing, Shenet al[39] demonstrated that obese and lean NAFLD-related HCCs in mice had a different mutational burden. For instance, they identified mutations in the carboxyl ester lipase gene that caused an increase in cholesterol esters mostly in the obese mice. Similarly, Grohmannet al[40] studied obese and lean mouse models to show that HCC and NASH development were dependent on divergent pathways, raising the possibility of variable mechanisms in non-cirrhotic HCC development. The non-fibrotic pathway contributions were also demonstrated in European cohorts (from Germany and the United Kingdom), in which polygenic risk scores (including PNPLA3, TM6SF2, membrane bound O-acetyltransferase domain containing 7, and glucokinase regulator) predicted the risk of HCC in patients with NAFLD. This risk was associated with hepatic steatosis (adjusted hazard ratio of 1.35,P< 0.01), even after correcting for hepatic fibrosis (P< 0.05)[41].
The advent of single cell RNA sequencing has allowed for further understanding of the cell type proportions in HCC, which was a limitation of bulk RNA sequencing given tumor heterogeneity[42], including the understanding of the inflammatory microenvironment that may have effects on treatment responses[43]. Whether similar cell type proportions and mutational signatures will be identified in NAFLD-related HCC remains to be seen in populations with and without cirrhosis.
A summary of the genetic variants and mutations described in NAFLD-related HCC is presented in Tables 1 and 2.
Epigenetic changes
Epigenetic modifiers also play a role in HCC development and account for approximately 32% of mutations found in HCC[44,45]. Many of the genes involved in structural chromosomal changes (AT-rich interaction domain 1A, AT-rich interaction domain 2, histone-lysine N-methyltransferase 2A) may not be directly involved in the pathogenesis of the disease but could be proxies to mutational changes in other genes linked by chromosomal looping captured by assay of transposase-accessible chromatin[46,47], an avenue that has not been yet explored in HCC related to NAFLD or to other etiologies of liver disease. Methylation aberrations also play a role. Recent work by Hernandez-Mezaet al[48] demonstrated the extensive methylation landscape of different etiologies of HCC in a European cohort, with a minority represented by NAFLD. Similar to the increase in mutational burden seen from normal liver to cirrhosis, the study demonstrated that patients with HCC were more likely to have hypermethylation patterns compared to controls. Interestingly, some of these differential methylation patterns involved key lipid genes, including the transcription factor, sterol regulatory element-binding protein 1.
Other factors
Serum metabolomic and microbiome studies have also identified signatures for poor NAFLD-related outcomes[49-51], although it remains to be seen whether these are surrogates for NASH progression or if they are involved in the pathways. The role of lipopolysaccharides has been studied in this context. The increase in lipopolysaccharides in NAFLD patients, as a surrogate for oxidative stress, is likely multifactorial and linked to the gut (bacterial overgrowth, increased permeability, among other factors), nutrients (including lipids), immune response, and hepatic injury, which adds another complexity to the NAFLD-related HCC spectrum of disease and potentially partly explains disease heterogeneity[52].
The use of metabolomics to identify signatures that are pathogenic in NAFLDrelated HCC is also a novelty in the field. A recent study by Buchardet al[53] aimed to identify differences in metabolomics in tissues of patients with NAFLD-related HCC by stratifying the cohort according to the degree of liver fibrosis. Using1H-nuclear magnetic resonance-based assays of 52 paired samples of HCC and adjacent nontumoral tissue, the authors identified that, independently of fibrosis stage, glucose metabolism was increased in tumors as were branched chain amino acids, potentially reflecting the activation of mechanistic target of rapamycin pathways, which parallels the genetic alternations of HCC discussed previously. This study also demonstrated that HCCs had lower levels of monounsaturated fatty acids, suggesting a lipid reprogramming in HCC. Similarly, HCCs developing in the setting of advanced fibrosis also had lower monounsaturated fatty acids compared to HCCs that originated in livers with no or mild fibrosis[53]. The differences observed in tumoralvsnon-tumoral tissues as well as in no or mild fibrosisvsadvanced fibrosis illustrate that tumorigenesis in NAFLD may have fibrosis-independent mechanisms as suggested by Grohmannet al[40]. On the other hand, most patients with NAFLD who develop HCC in the absence of cirrhosis have NASH and advanced liver fibrosis instead of simple fatty liver with no or mild fibrosis, which could imply an association between fibrosis and hepatocarcinogenesis as well as common mechanisms for NASH and NAFLDrelated HCC[12]. In this regard, the lipotoxicity and the metabolic reprogramming associated with steatosis are examples of pathogenic factors involved in the development of both NASH and HCC, and the inflammatory microenvironment of NASH also favors hepatocarcinogenesis[3].
Other genetic alterations that are a focus of current interest in NAFLD-related HCC are non-coding RNAs. Depending on further studies, they may provide an additional layer of complexity in epigenetic changes[45].
IMMUNE ASPECTS OF NAFLD-RELATED HCC
The mechanisms underlying the initiation and progression of HCC in the background of NAFLD are not fully understood. A number of factors including hepatic lipotoxicity, chronic inflammation, progressive fibrosis, and changes in the microbiome have all been implicated in NAFLD-related hepatocarcinogenesis. Recent studies have elegantly elucidated the role of the tumor microenvironment in thisscenario[3,54-57]. Moreover, other authors have comprehensively discussed the role of cancer cell intrinsic factors that drive HCC in NAFLD[3,54,58,59]. Nevertheless, the role of the host immune system in NAFLD-related hepatocarcinogenesis must also be highlighted.

Table 2 Summary of genetic mutations described in nonalcoholic fatty liver disease-related hepatocellular carcinoma
The liver is considered an immunologically privileged organ. It is constantly exposed to metabolites, toxins, and microbial products from the intestine since it derives a large part of its blood supply from the portal vein. However, there are several immune mechanisms within the liver that prevent an inflammatory hyperresponse to this physiological antigenic load, including reduced expression of major histocompatibility class proteins, suppressed antigen presentation by Kupffer cells and dendritic cells, and enrichment of immunosuppressive cells like the regulatory T cells[60-62]. These mechanisms are overwhelmed in the context of NAFLD, where progressive steatosis leads to lipotoxicity, mitochondrial dysfunction, oxidative stress, and activation of cell death pathways, all of which trigger a state of chronic sterile inflammation. Unfortunately, a combination of the same factors that drive NASH progression also play mechanistic roles in the initiation of HCC in the background of this inflammatory milieu.
Progressive NASH influences both the innate and adaptive arms of the immune system, which together can enable cancer initiation and progression. The complex crosstalk among hepatocytes, adaptive immune cells, and cancer cells has been demonstrated by several studies. Wolfet al[54] found that infiltrating CD8+ T cells and natural killer cells contribute to NASH development and the subsequent transition to HCC. However, another study using a different mouse model of NASH showed that CD8+ T cells prevented HCC development and that a specific subset of immunosuppressive IgA+ plasma cells expressing programmed cell death ligand-1 and interleukin-10, which were abundant in NASH livers, directly suppressed liver cytotoxic CD8+ T cells, leading to HCC development[56]. Subsequently, Maet al[55] showed that the metabolic dysregulation in NAFLD causes selective loss of CD4+ T lymphocytes, thus contributing to accelerated hepatocarcinogenesis. Meanwhile, Gomeset al[57] have shown that T helper 17 cells are activated upon hepatocyte DNA damage in NASH and can promote HCC.
Innate immune cells like macrophages, dendritic cells and natural killer cells are also important in the pathogenesis of NAFLD-related HCC. Kupffer cells are resident macrophages that play a significant proinflammatory and profibrotic role during NASH progression. However, their role in HCC is not clear yet. Wuet al[63] showed that the activation of Kupffer cells positive for triggering receptor expressed on myeloid cells-1 led to secretion of proinflammatory cytokines like interleukin-6, interleukin-1β, tumor necrosis factor, C-C motif chemokine ligand 2, and C-X-C motif chemokine ligand 10, which in turn promoted HCC. In general, though, protumorigenic M2-like macrophages that drive tumor progressionviasuppressing cytotoxic T cells and inducing angiogenesis appear to be recruited from circulating bone marrow derived monocytes rather than resident macrophages[64,65]. Other immune cells like neutrophils[66-68], monocytes[69], dendritic cells[70], and natural killer cells[71,72] have also been implicated in HCC progression in NASH, highlighting the complexity of the immune mechanisms of NAFLD-related hepatocarcinogenesis (Figure 2).
HCC IN NAFLD WITHOUT CIRRHOSIS
Given some of the specificities involved in NAFLD-related hepatocarcinogenesis, HCC in the setting of NAFLD is known to occur even in the absence of liver cirrhosis, an event previously related mostly to hepatitis B virus infection[12]. The prevalence of NAFLD-related HCC in the absence of cirrhosis varies dramatically according to the geographic location of the study and even among different studies performed in a similar region of the world. Most experts estimate that between 14% and 54% of NAFLD-related HCC cases occur in patients without cirrhosis. A study from the Veterans Affairs (VA) Health System in the United States by Mittalet al[73] found that 42% of veterans with NAFLD-related HCC had no evidence of cirrhosis. Interestingly, a similar study by the same group the following year found the prevalence of noncirrhotic HCC related to NAFLD to be 13%[74]. In the latter study, however, the estimation of cirrhosis was separated by different levels of confidence. Small studies from Italy and Japan have also found that 50% and 48% of NAFLD-related HCC cases, respectively, occurred in the absence of cirrhosis, suggesting that the burden of noncirrhotic HCC in NAFLD is also significant in other parts of the world[75,76]. Finally, a meta-analysis of 19 studies found the prevalence of non-cirrhotic HCC among NAFLD-related HCC to be approximately 38%[77].
Several issues help explain the variable results from multiple studies: (1) classifying patients as to whether or not they have cirrhosis through liver biopsy is possible mainly in small studies, while this classification is much less precise in larger studies that look at International Classification of Diseases codes or large commercial clinical databases; (2) most studies in the United States have been performed in the VA System, which is inevitably biased towards a large presence of male gender among the evaluated cohorts (> 90% in most studies[32,73,74,78]); and (3) the distinction between NAFLD and NASH is not completely clear in all the studies. In this regard, a study from the Netherlands looking at almost 100 non-cirrhotic NAFLD-related HCC cases found that most individuals had a low degree of or no steatohepatitis at all, suggesting a non-inflammatory carcinogenesis path towards HCC in this setting[79].
The lack of clarity on mechanisms leading to non-cirrhotic HCC with underlying NAFLD presents a difficult dilemma for practicing providers, as it is unclear who to screen for HCC. A retrospective cohort study of 271906 patients from the VA System (mean body mass index of 31.6 kg/m2, 28.7% with diabetes, 70.3% with hypertension, 62.3% with hyperlipidemia) suggested that diabetes and hyperlipidemia increase the risk of HCC in NAFLD[80]. However, the overall proportion of people with diabetes and NAFLD is still elevated as a total number of individuals to screen. Indeed, between 40% to 70% of individuals with diabetes have evidence of NAFLD[81]. Furthermore, it is unclear if the correlation between diabetes and HCC in patients without cirrhosis applies to other populations, as a recent study from Europe, characterizing the differences between cirrhotic and non-cirrhotic HCC in NAFLD, found an inverse association between diabetes and HCC in the non-cirrhotic group. Interestingly, non-cirrhotic HCCs in this study tended to occur in older patients and with lower body mass index[82]. As described below, the understanding of how to surveil patients with NAFLD for HCC is in its infancy, and further studies are needed to better define those at risk.

Figure 2 Main immune mechanisms of nonalcoholic fatty liver disease-related hepatocarcinogenesis.
SURVEILLANCE FOR HCC IN NAFLD
Surveillance programs aim at allowing for early detection of HCC among high-risk patients so that they have higher odds of being candidates for curative treatments. In fact, when HCC is diagnosed during surveillance, it is diagnosed in earlier stages[83-86], and patients have significantly higher survival rates[85,87]. Thus, it is of utmost importance to define which patients should be submitted to surveillance.
For individuals with an estimated annual incidence of HCC ≥ 1.5%, surveillance is considered cost-effective[8], but it is not always clear which subgroups of patients reach such a cutoff. The main risk factor for HCC in patients with NAFLD is cirrhosis, and therefore the most important international guidelines are consensual that individuals with NAFLD and cirrhosis should be surveilled for HCC with ultrasonography (US) every 6 mo[88-91]. It should be highlighted, though, that obesity and steatosis might impair the performance of US[8], and the American Gastroenterological Association recommends using either computed tomography scan or magnetic resonance imaging in cases in which US quality is deemed unacceptable[91]. Regarding the use of biomarkers, some guidelines make it optional to add alphafetoprotein to the surveillance program[89-91], but its performance is suboptimal, especially in NAFLD-related HCC[8], and new biomarkers should be pursued, such as those currently under study by the European-South American Consortium to Assess Liver-Originated Neoplasia.
Despite these recommendations, patients with NAFLD-related cirrhosis seem to be less likely to undergo surveillance than those with other underlying liver diseases[86,92]. In order to overcome the low adherence to surveillance, screening tools to identify individuals at higher risk for HCC could be useful. The GALAD score (gender, age, lectin-binding alpha-fetoprotein-3, alpha-fetoprotein, and des-gamma-carboxyprothrombin) has been studied in this context, and it has been recently validated in patients with NASH. In such patients, the GALAD score had sensitivity and specificity over 90% to identify individuals who would develop HCC as early as 1.5 years before the diagnosis[93].
However, some authors believe that in order to stratify patients according to their risk of developing HCC, different tools might be necessary depending on the underlying liver disease. Using data from the VA Health System database, a study evaluated 7068 patients with NAFLD and cirrhosis, with an annual incidence of HCC of 1.56%. A predictive model based on age, sex, platelet count, albumin levels, aspartate aminotransferase/alanine aminotransferase ratio, diabetes, and body mass index was developed, and it had an area under the receiver operating characteristic curve of 0.775 and 0.721 for predicting HCC in the derivation- and in the validationcohorts, respectively. This model was able to classify patients as low-risk (< 1%/year), medium-risk (1%-3%/year), and high-risk (> 3%/year) for HCC. A classification such as this could be used, if further validated, to define subgroups that might spare surveillance[78].
As discussed above, there are subgroups of patients with NAFLD who do not have cirrhosis but are at risk of developing HCC. In a large retrospective cohort study including 296707 individuals with NAFLD and a similar number of matched controls from the VA Health System database, patients with NAFLD had 7.6-fold higher risk of developing HCC than their counterparts, and the risk was greater among men, older people, and Hispanics. However, in the NAFLD-group, the annual incidence of HCC was 10.6/1000 person-years for individuals with cirrhosis and 0.08/1000 person-years for those without it, which was considered insufficient for a general recommendation of surveillance to be made for patients without cirrhosis. The FIB-4 score was also evaluated, and, despite its association with the development of HCC, individuals with high FIB-4 scores (> 2.67) but without a diagnosis of cirrhosis were still considered to have a low risk of developing HCC[32].
Another large study evaluated four European primary care databases including over 18 million individuals and verified an incidence of HCC of 0.3/1000 person-years among patients with NAFLD, which was much higher than that of controls (hazard ratio of 3.51). When the NAFLD group was classified according to the FIB-4 score, it was possible to identify which patients were under higher risks. When compared to individuals with a FIB-4 score < 1.30, those with scores between 1.30 and 2.67 had a hazard ratio for HCC of 3.74, and the ones with scores > 2.67 had a hazard ratio of 25.2[94]. Therefore, despite conflicting evidence, it is possible that the FIB-4 score could be used in order to select patients for surveillance.
Currently, guidelines are vague regarding surveillance for HCC in patients with NAFLD who do not have cirrhosis. The American Gastroenterological Association, in its position paper on surveillance for HCC in patients with NAFLD, recommends considering patients with NAFLD and advanced fibrosis for surveillance but recommends against routinely surveilling individuals with earlier stages of fibrosis[91]. While the position of the European Association for the Study of the Liver is similar to that[88], the American Association for the Study of Liver Diseases considers the benefit of surveillance in individuals with NAFLD who do not have cirrhosis to be uncertain and does not support it[90].
DISCUSSION
NAFLD currently affects one fourth of the global population[2]. Its increasing prevalence and the fact that it is associated with the development of liver cancer, both in the setting of cirrhosis and in its absence, make NAFLD-related HCC a growing challenge[12]. It is likely that the growth in NAFLD-related HCC will offset a decrease in viral hepatitis-related liver cancer, which is expected for the near future due to vaccination against hepatitis B virus and to the highly effective treatments for hepatitis B and C[95]. NAFLD-related HCC is already responsible for an important burden on public health, being associated with 796000 disability-adjusted life years in 2019, an increase of 33.6%in comparison to 2010[4].
This article has highlighted important genetic and immune-mediated mechanisms involved in NAFLD-related hepatocarcinogenesis. Understanding the role of certain genetic variants (especially those associated with genes such asPNPLA3[22,23],TM6SF2[24,25], and membrane bound O-acetyltransferase domain containing 7[25-30]) as well as the importance of epigenetic modifiers[44,45], the microenvironment of NAFLD, and the influences that this disease has on the innate and adaptive immune systems[54-57] will hopefully allow for a better knowledge of the clinical characteristics of NAFLD-related HCC, including the possibility of the development of liver cancer in the absence of cirrhosis. Moreover, this knowledge may help define more appropriate surveillance strategies, focusing not only in individuals with cirrhosis, since over one third of NAFLD-related HCC cases are diagnosed in patients without this condition[77]. At present, surveillance with US every 6 mo is recommended for individuals with advanced liver fibrosis[91].
This review has limitations associated especially with the incomplete understanding of NAFLD-related HCC by the scientific community. The pathophysiology of this condition must be further studied, particularly the mechanisms leading to noncirrhotic HCC. Moreover, there is a profound necessity for the identification of better biomarkers to detect subgroups of patients that could benefit from surveillance aside from those with cirrhosis[96].
CONCLUSION
The worldwide growing prevalence of NAFLD and its association with the development of HCC in patients either with or without cirrhosis make NAFLD-related HCC a growing challenge. Improving surveillance strategies is of the utmost importance in order for the early detection of HCC and for patients to have higher chances of being cured. Further understanding of the mechanisms leading to HCC in the setting of NAFLD will likely lead to novel molecular candidates that could be used as biomarkers to identify patients who will progress to develop a liver malignancy even in the absence of cirrhosis.
杂志排行

World Journal of Hepatology的其它文章
- Coronavirus disease 2019 and non-alcoholic fatty liver disease
- Epigenetic mechanisms of liver tumor resistance to immunotherapy
- Advances in the management of cholangiocarcinoma
- Herbal and dietary supplement induced liver injury: Highlights from the recent literature
- Challenges in the discontinuation of chronic hepatitis B antiviral agents
- Liver Kidney Crosstalk: Hepatorenal Syndrome