Epigenetic mechanisms of liver tumor resistance to immunotherapy
2021-10-11JulieSanceauAngliqueGougelet
Julie Sanceau, Angélique Gougelet
Julie Sanceau, Angélique Gougelet, Centre de Recherche des Cordeliers, Sorbonne Université, Inserm, Université de Paris, Paris 75006, France
Abstract Hepatocellular carcinoma (HCC) is the most common primary liver tumor, which stands fourth in rank of cancer-related deaths worldwide. The incidence of HCC is constantly increasing in correlation with the epidemic in diabetes and obesity, arguing for an urgent need for new treatments for this lethal cancer refractory to conventional treatments. HCC is the paradigm of inflammation-associated cancer, since more than 80% of HCC emerge consecutively to cirrhosis associated with a vast remodeling of liver microenvironment. In the recent decade, immunomodulatory drugs have been developed and have given impressive results in melanoma and later in several other cancers. In the present review, we will discuss the recent advancements concerning the use of immunotherapies in HCC, in particular those targeting immune checkpoints, used alone or in combination with other anticancers agents. We will address why these drugs demonstrate unsatisfactory results in a high proportion of liver cancers and the mechanisms of resistance developed by HCC to evade immune response with a focus on the epigeneticrelated mechanisms.
Key Words: Liver cancer; Immunotherapies; Epigenetics; Resistance; Hepatocellular carcinoma
INTRODUCTION
Hepatocellular carcinoma (HCC) is the most common primary liver tumor with 800000 newly diagnosed people per year in the world[1]. HCC also stands fourth in rank of deaths related to cancer worldwide, accounting for more than 700000 deaths per year. Liver cancer incidence has tripled since the 80s and reaches a high incidence in western countries consequently to obesity and diabetes epidemic, supporting the need of novel effective strategies for this cancer refractory to the majority of conventional anticancer treatments. HCC is a complex disease but its mutational landscape has been extensively uncovered these two last decades with advances in deep-sequencing technologies. The most recurrent mutations identified in HCC are mutations inTERT,CTNNB1andTP53[2], but other frequent mutations in epigenetic modifiers and chromatin remodelers are also encountered (e.g.,ARID1A, ARID2, MLL2)[3,4]. Other crucial epigenetic modulators, the non-coding RNAs (ncRNAs), are also largely deregulated during hepatocarcinogenesis, reprogramming tumor cells but also modifying the surrounding cells and secondary sites of metastasisviatheir secretion[5].
Integrating outside and inside signals in time and space, the epigenetic regulations of gene expression is a crucial determinant of tumor cell fate regarding differentiation, proliferation, metabolism, migration and immunosurveillance. Epigenetic modifications are categorized into three main mechanisms: DNA methylation, histone modifications mainly on H3 and H4 histones (acetylation, methylation,etc.) and control by ncRNAs. There is a growing body of evidence that epigenetic modifiers play key roles during cancer, including in HCC. Therefore, they constitute attractive therapeutic options, alone or in combination with other anti-cancer agents, such as drugs targeting DNA methylation and histone acetylation, which have already been approved for hematological cancers[6]. These recent years, it has been extensively documented that the immune response is epigenetically controlled and plays critical roles in tumor immunosurveillance. Among others, epigenetic changes impact macrophage polarization, myeloid-derived suppressor cell (MDSC) function, genesis of cancer-associated fibroblasts and function of T cell populations, either CD4+, CD8+ and T regulators (Tregs). Of note, subsets of inflammatory gene promoters have been found epigenetically deregulated in cancer. In particular, aberrant DNA methylation of interferon-γ (IFNγ) is associated with exhausted phenotype of T cells[7]. The cytokines involved in THresponse have been found epigenetically inhibited by EZH2 (Enhancer of zeste homolog 2) and DNMT1 (DNA methyltransferase 1)[8]-infiltration of CD8+ cells being inversely associated with the high expression of EZH2. In addition to cytokines, the expression of immune checkpoints such as the program cell death 1 (PD-1)/program cell death ligand 1 (PD-L1) axis is also regulated by epigenetic modifications. DNA methylation in the promoter region ofCD274encoding PD-L1 predicts patient survival in multiple cancers. EZH2 modifies its H3K27 trimethylation status in hepatoma cells[9], while the BET protein BRD4 (bromodomain-containing protein 4), found overexpressed in HCC and enriched on super-enhancers driving oncogene expression[10], suppressed PD-L1 expression[11].
HCC is the paradigm of inflammation-associated cancer, since more than 80% of HCC emerge consecutively to cirrhosis associated with a vast remodeling of liver microenvironment. Immune cell remodeling is a consequence of chronic hepatitis or liver disease associated with alcohol consumption, genotoxic exposure or metabolic disorders[12]. Even if liver parenchyma harbors a specialized and protective immune system to manage its constant exposure to toxins and bacteria susceptible to trigger deleterious inflammation, the chronicity of hepatic injuries sensitizes to HCC. In liver cancers, as in a number of other cancers, tumor microenvironment differs accordingly to the driven oncogenic mutations and thus impacts response to treatments, notably to immunomodulatory drugs[13]. Cancers withCTNNB1mutations have been defined as cold tumors with lower immune cell infiltration and refractoriness to immune checkpoint inhibitors (ICIs)[14,15]. Indeed, the Wnt/β-catenin pathway plays a major role in the specification of a multitude of immune cells including macrophages, dendritic cells (DC) and lymphocytes[16].
In the present review, we will discuss the recent advances on immunotherapies in clinical practice, successfully used alone or in combination with other anti-cancers agents in several cancers. We will also address why these drugs demonstrate unsatisfactory results in a high proportion of liver cancers, which shown innate or acquired resistance to immunomodulatory agents. We will thus detail the mechanisms of resistance developed by HCC and particularly the epigenetic-related mechanisms.
MECHANISMS OF T CELL ACTIVATION AND ATTENUATION
T cell activation needs two signals from antigen presenting cells (APC). The initial signal is based on antigen recognition through interaction between T cell receptor (TCR) complexed to CD3 subunits on T lymphocytes and its cognate antigen/MHC (major histocompatibility complex) on APC (Figure 1). This interaction promotes CD3 phosphorylation on ITAM motifs (immunoreceptor tyrosine-based activation motifs) which serve as docking sites for the recruitment of ZAP-70 (TCR-ζ chain-associated 70-kDa tyrosine phosphoprotein) and subsequent phosphorylation by Lck (lymphocytespecific protein tyrosine kinase) and autophosphorylation. Once fully activated ZAP-70 phosphorylates LAT (linker of activated T cells) and SLP-76 (SH2 domaincontaining leukocyte protein of 76 kDa), two adaptors for the assembly of the complete TCR signalosome. Secondary signals are required to fully activate LAT. The costimulatory signals are mostly provided by members of the immunoglobulin superfamily such as CD80(B7-1)-CD86(B7-2) bound to CD28, ICOSL to ICOS (inducible T-cell costimulator) (respectively on APC and T cell), or those of the tumor necrosis factor (TNF) receptor superfamily (e.g., OX40L-OX40, CD40/CD40L).
To avoid excessive immune response, co-inhibitory molecules, including CTLA-4 (cytotoxic T lymphocyte antigen 4), PD-1 and LAG-3 (lymphocyte-activation gene 3), act as negative immune counterweights (Figure 1). Inhibitory receptors mediate their negative regulation through inhibitory motifs located in their cytoplasmic tails such as immunoreceptor-based inhibitory motif (ITIM) to recruit phosphatases containing Src homology-2 domains, such as SHP-1 and SHP-2 (small heterodimer partner). The recruited phosphatases dephosphorylate several molecules involved in the TCR signaling such as the TCR itself or ZAP-70. This interrupts downstream cascades such as the PI3K (phosphoinositide-3-kinase)/AKT and the rat sarcoma virus (Ras)/rapidly accelerated fibrosarcoma (Raf)/mitogen activated protein kinase kinase (MEK)/ extracellular signal regulated kinase (ERK) and leads to reduction in T cell activation, proliferation, metabolism, differentiation, survival, and cytokine production. In addition, PD-1 as well as CTLA-4 are also able to directly regulate signaling pathways in lymphocytes such as the PI3K and MAP kinase pathways[17-19]. While CTLA-4 is the leading player of the ICIs limiting priming of naive T cells notably in lymph nodes, PD-1/PD-L1 interaction results in exhaustion of activated T cells in peripheral tissues and within the tumor microenvironment.
PD-1/PD-L1 axis
PD-1, also known as CD279, is low or undetectable in naive T cells and rapidly induced following TCR activation, in a process partially regulated by transforming growth factors β (TGF-β)[20]. PD-1 is also expressed on other several cells such as B lymphocytes, natural killer (NK), macrophages, DC and monocytes and tumor-specific T cells. At the transcriptional level, PD-1 expression is regulated by nuclear factor of activated T-cells (NFAT)[21], forkhead box O (FOXO)[22] and interferon regulatory factor 9 (IRF9)[23], STAT3/4 (signal transducer and activator of transcription 3 and 4) and CTCF (CCCTC- binding factor)[24] (Figure 2). PD-1 content is also dependent on microRNAs (miRNAs) such as miR-28[25], miR-138 and miR-4717 in glioma[26] and HCC respectively[27]. Differential level of the repressive H3K9me3 mark has been observed in the promoter region of PD-1 in colorectal cancer[28].
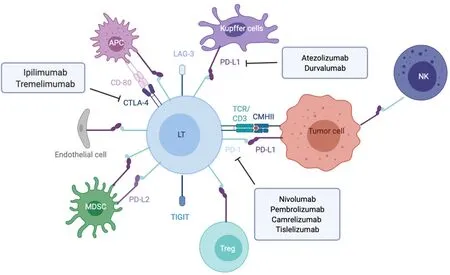
Figure 1 Overview of the main immune checkpoint and their respective targeted therapies.
PD-1 triggers immunosuppressive signals upon binding to its ligands, PD-L1 (CD274 or B7-H1) and PD-L2 (CD273). A soluble form of PD-L1 (sPD-L1) is secreted in the blood and could compete for PD-1 binding with membranous PD-L1. PD-L2 is restricted to APCs and B lymphocytes, while PD-L1 is usually expressed by macrophages, DC, epithelial cells, activated T cells and B cells. To escape anti-tumor response, PD-L1 expression is highly induced in tumor cells. This could result from genomic alterations such as amplification of translocation including in HCC[29]. Gain in PD-L1 copy number is also a frequent alteration across many cancers, which influences PD-L1 expression levels and correlates with higher number of mutated genes[30]. Nevertheless, such a correlation is not observed in HCC.CD274expression is controlled by DNA methylation and could constitute a prognosis factor in colon[31] or prostate cancers[32]. Several signaling pathways are also well documented to induce PD-L1 expression in tumor microenvironment such as interferon signaling, PI3K-AKT, MEK-ERK, JAK-STAT, c-MYC and NF-kB (nuclear factor-kappa B)[33]. This transcriptional regulation is regulated by a plethora of cytokines and growth factors such as IFN-γ, interleukin (IL)-6, IL-17, IL-25, TNF-α or epidermal growth factor (EGF)[34]. PD-L1 expression is also regulated by several miRNAs found implicated in cancers: miR-15/miR-16/miR-193a[35], miR-17[36], miR-34[37], the miR-25/miR-93/miR-106b cluster[38], miR-138-5p[39], miR-140[40], miR-142-5p[41], miR-152[42], miR-197[43], miR-200[44], miR-217[45], miR-324-5p/miR-338-5p[46], miR-424[47], miR-513[48], and miR-570 in HCC[49].
CTLA4/CD80-CD86 axis
CTLA-4 is a CD28 homolog which interacts with CD80 and CD86 with higher affinity and avidity than CD28. Therefore, CTLA-4 enters in competition and prevents the stimulatory signals induced by CD28:CD80/CD86 complexes. Membranous CTLA-4 expression is very low in resting T cells, consequently to clathrin-dependent recycling, and increases following T-cell activation[50]. CTLA-4 is thus mostly localized in intracellular compartments such as lysosomal and endosomal vesicles and the trans Golgi network. CTLA-4 expression is also regulated at the transcriptional level by NFAT[51]. Importantly, CTLA-4 expression has also been detected on tumor cells, including melanoma, colon and renal cancers[52]. In cancer cells, notably in melanoma, CTLA-4 expression is regulated by IFN-γ signaling pathway and DNA methylation[53] but also induced by β-catenin binding on a lymphoid enhancer factor-1 (LEF-1) binding site in its promoter region[54]. In line with these regulations, theCTLA4gene displays several SNPs (single-nucleotide polymorphism) associated with disease and cancer in its promoter as well as in its first exon. In particular, theCTLA-4 318C > TSNP creates a LEF-1 binding site in its promoter and increase CTLA-4 expression and antitumor activity[55]. CTLA-4 expression is also epigenetically regulated with lower level of repressive H3K27me3 mark detected in CTLA-4 promoter in colorectal cancers[28]. CTLA-4 expression is also post-transcriptionally regulated by miR-9/miR-155[56], miR-138[26] and miR-487a-3p[57].
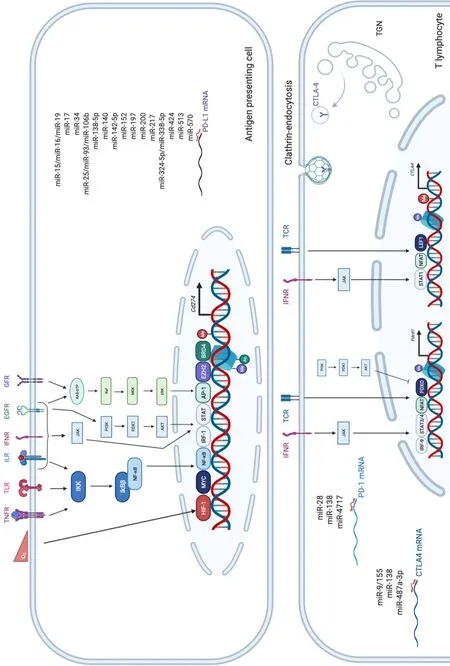
Figure 2 Overview of the main epigenetic and transcriptional regulations of program cell death 1, program cell death ligand 1 and cytotoxic T lymphocyte antigen 4.
Regarding CTLA-4 ligands, contrary to PD-L1, CD80 and CD86 are restricted to lymphoid cells. While CD80 is generally poorly detected on resting cells and upregulated after activating signals, CD86 is ubiquitously expressed on DCs, monocytes and activated B cells and induced at high levels upon activation. The regulation of these molecules is less detailed. In DCs, CD80 expression is reduced in response to miR-424[47]. Low levels of CD80 and CD86 have been detected on melanoma and colon cancer cells, where low level of CD80 expression favors tumor growth[58] but also on HCC cells, as shown by a pioneer study supporting the potential of CTLA-4 axis targeting as anticancer therapy[59].
MECHANISMS OF IMMUNE ESCAPE AND IMMUNOTHERAPY
The goal of immunotherapies is to boost ability of the immune system to detect tumors and limit their progression. They might counteract the evasion mechanisms mediated by the suppressive molecules rolled out by tumor cells. Different therapeutic strategies have been developed but ICIs, designed to block the co-inhibitory signals of T-cell activation (e.g., CTLA-4, PD-1 and PD-L1), are the preferred methods in clinical practice. These drugs have given very impressive results with cancers of bad prognosis and with few therapeutic options, such as melanoma, and have been rapidly tested in several other tumors with high clinical efficacy in most cases.
Mechanisms of tumor immune evasion
Tumor development and progression is a complex process resulting from the interplay between cancer cells and its surrounding environment including endothelial cells, fibroblasts, and a plethora of immune cells with suppressive, regulatory, killing and either anti or pro-inflammatory functions. All types of immune cells are present in the tumor or in the invasive margin, including macrophages, DCs, mast cells, NK cells, naive and memory lymphocytes, B cells, and effector T cells (e.g., Th1, Th2, Th17, Treg and cytotoxic T cells). Therefore, the strength of anti-tumor immune response is governed by the level and the composition of immune cell infiltrated in the tumors and the degree of T cell activation.
As previously mentioned, tumor cells are able to express co-inhibitory ligands such as PD-L1 or PD-L2, and sometimes inhibitory receptors such as PD-1 including in HCC[60,61]. This prevents T cell activation and modulates the activity of recruited immune cells, which express the cognate molecules and play suppressive activities such as tumor-associated macrophages (TAM), myeloid-derived suppressive cells or Tregs[62] (Figure 3). Accumulation of suppressive cells and T dysfunction are also sustained by several molecules secreted by tumor cells such as PGE2 (prostaglandin E2), COX2 (cyclooxygenase 2), nitric oxide, TGF-β and IL-10[63]. Additionally, multiple cancers are associated with chronic inflammation, particularly HCC related to hepatitis infection. Chronic disease results in an ineffective T response and T cell exhaustion mostly due to persistent inflammatory signals, antigen exposure and suppressive cytokines such as IL-10 and TGF-β. It has also been described that chronic disease modifies PD-1 promoter status in exhausted T cells that remains demethylated and poised to facilitate its rapid expression[64,65]. Progressively, exhausted T cells lose their proliferative capacity and effector function related to decrease in IL-2, TNF-α and IFN-γ.
Tumor cells are also able to modify T cell expansion through metabolic alterations. In particular, an overexpression of IDO (indoleamine-2,3-dioxygenase), an enzyme involved in tryptophan conversion, is frequently observed in tumors[66] as well as overexpression in arginase, particularly in MDSC[67]. The depletion of tryptophan and arginine in tumor microenvironment reduces T cell proliferation[68,69].
Tumor immune privilege is also the consequence of decrease in the expression of recognition molecules including MHC, tumor-associated antigens (TAA) and tumorspecific antigens. It is well described that changes in antigens expressed by tumor cells are detected by the immune system, which further develop autoantibodies against TAAs as reporters to control the transformation process. The typical antigen with autoantibodies identified in cancer is p53[70]. Antigens in HCC could be categorized from cancer testis origin such as SSX-2 (synovial sarcoma, X breakpoint 2) and MAGE (melanoma antigen gene), or oncofetal antigens such as α-fetoprotein and glypican 3 or overexpressed tumor antigens such as annexin A2 and epithelial cell adhesion molecule. They constitute promising targets for adoptive cell therapies such as chimeric antigen receptor T cells or tumor-infiltrating lymphocytes (TILs)[71]. A higher expression of TAAs in HCC patients is correlated with higher immune infiltration and better prognosis[72]. The loss or modification of antigens promote immune evasionviaa defect of tumor recognition. Shedding of natural killer group 2D (NKG2D) ligands into the tumor microenvironment is another way to evade immune recognition. Following proteolysis by matrix metalloproteinases, tumor cell death or exosome secretion, the soluble form of NKG2D ligand induces internalization and degradation of NKG2D and decrease the subsequent cytotoxic effects of T cells[73].
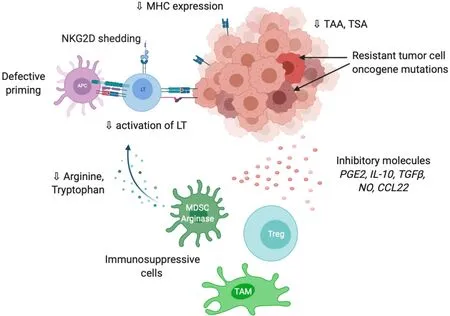
Figure 3 Overview of the main mechanisms involved in tumor evasion to immune response.
Independently from tumor microenvironment, tumor cells resist to destruction through additional mutations in oncogenes (BRAF, EGFR, HER2,etc.) that give proliferative advantage. Inversely, mutations in tumor suppressive molecules in particular in damage sensors and pro-apoptotic actors (TP53, BCL2,etc.) also limits the cytotoxic activity of the immune system[74].
Tumor-infiltrating immune cells
Tumor immune response and subsequent efficacy of ICI treatment is also highly dependent on the immune cell spectrum and its localization within or around the tumors. Indeed, pathological characterization of various solid tumors has shown a great diversity in immune cell types and density between tumors, which could be dependent on driver oncogenes. Three groups have been characterized either as immune desert, immune excluded or inflamed tumors – each group being associated with differential response to ICIs[75].
The inflamed tumors are characterized by the presence of CD8+ and CD4+ T cells with suppressive cells including macrophages, MDSC and Treg that promote T cell dysfunction and exhaustion[76]. In immune-excluded tumors, aggregates of immune cells are at the tumor boundaries. Immune cells are not recruited in the vicinity of tumors consequently to physical hindrance associated with dense and stiff extracellular matrix fibers, defect in neo-vasculature, hypoxia, low level of chemoattractive molecules for T cells such as C-X-C motif chemokine ligand 9 (CXCL9) and CXCL10, insufficient level of antigens or exposure to microbes or virus. In immune desert or cold tumors, there is a low density of immune cells inside and outside the tumors. Tregs, MDSCs and macrophages interplay to inhibit DC maturation and impair T cell expansion and activation. Growing body of data have shown that EMT (epithelial-to-mesenchymal transition) and mesenchymal traits of tumor cells favor immune exclusion and resistance to ICIs[77].
In 2017, a new molecular HCC classification has been proposed on the basis of immune traits, with approximately 30% of HCCs enriched in TILs and defined as HCC immune class[15]. Thirty percent of patients inversely showed exclusion of TILs and frequent mutations inCTNNB1gene. This subgroup of tumors are resistant in firstintention to ICIs[13], as it was previously observed in melanoma[78]. This was confirmed with a hydrodynamic mouse model of HCC in which β-catenin activation promotes immune evasion and resistance to anti-PD-1 therapy[79].
In addition to CD8 T cells, the distribution pattern of myeloid cells has also been associated with HCC prognosis. A recent work of Wu and collaborators proposed a myeloid response score (MRS) associated with T cell activity and which could serve as a prognosis signature[80]. HCC were classified as HCCs with low, intermediate, and high MRS, which displayed patterns of immunocompetent, immunodeficient, and immunosuppressive microenvironment. MRSlowtumors present an intratumor contexture equivalent to the peritumor tissue containing CD169+CD163+CD14+CD11blow/-macrophages with antitumor activity and CD8+ T cells. Inversely, as compared to nontumor tissue MRShightumors are enriched in CD11b+CD15+ polymorphonuclear leukocytes and CD169-CD11b+CD163+ myeloid cells associated with pro-tumoral activation of TAM. These tumors are also characterized by gene signatures related to immunosuppression.
The expression of co-inhibitory molecules within the tumor is an important prognosis factor. HCC with high expression of PD-L1 on tumor/immune cells in immunohistochemistry together with high expression of PD-1 on lymphocytes also exhibit markers of aggressiveness such as poor differentiation and vascular invasion[81]. In addition, if PD-L1 is overexpressed by HCC cells, this predicts early recurrence. Importantly, in this study, no correlation between glutamine synthetase, a direct positive target of the β-catenin, and PD-L1 labeling was observed meaning that the immunosuppressive activity of the Wnt/β-catenin could thus be linked to an immune checkpoint other than PD-L1/PD-1 axis. Another study performing cytometry analysis on HCC tumors confirmed that PD-L1 was both expressed by tumor cells and immune cells and mostly on CD68+ myeloid cells[82]. The presence of PD-L1 on tumor cells correlates with tumor progression, while PD-L1+ macrophages play a protective role in HCC associated with immune response and T activation signature. Recently, a TCGA analysis showed that a high correlation between all negative checkpoints such as PD-L1, PD-1, CTLA-4, LAG-3 and T infiltration in tumors is associated with an immunosuppressive and exhausted tumor microenvironment[83]. Nevertheless, the application of ICIs would be of survival benefit for these patients.
IMMUNOTHERAPY SUCCESSES AND LIMITATIONS IN HCC
Development of immune checkpoints inhibitors constitutes a major breakthrough in oncology that leads to revisit therapeutic strategies and clinical practice for various cancers particularly those of poor prognosis with few therapeutic options, following impressive results obtained in melanoma. ICIs have resulted in increased patient survival in melanoma, kidney and non-small cell lung cancer as well as Hodgkin’s lymphoma in comparison with conventional chemotherapies. Other cancers present a more heterogenous response to ICIs such as ovarian, breast, pancreatic and liver cancers. More promising data have been obtained with combination of treatments including ICIs. Microsatellite instability has been evidenced as a biomarker for ICI response[84]-tumors with a low mutation rate having less neoantigens and thus being less immunogenic. Another biomarker is TMB (Tumor mutational burden) has been recently found correlated with ICI sensitivity[85].
Anti-CTLA-4 therapy is the first generation of ICI since antitumor regression after blocking co-inhibitory molecules was firstly evidenced with the anti-CTLA-4 antibody ipilimumab in melanoma[86]. It was the first ICI approved by the Food and Drug Administration (FDA) for the treatment of advanced melanoma. Therapeutic strategies against PD-1 are the second generation of ICI with nivolumab and pembrolizumab lately approved by FDA for advanced melanoma[87]. Since then, the impacts of both therapies have been explored in various cancers and several others surface molecules have been targeted: Inhibitory co-receptors such as VISTA (V-domain Ig suppressor of T cell activation)[88], TIGIT (T Cell Immunoreceptor With Ig And ITIM Domains)[89], TIM-3 (T cell immunoglobulin and mucin domain-containing protein 3)[90] and LAG-3[91] or costimulatory receptors like CD28, OX40[92] or GITR (glucocorticoid-induced TNFR-related protein)[93].
Ipilimumab was the first blocking antibody to significantly promote a regression of lesions in metastatic melanoma with a complete remission in some patients[94]. A 3-year overall survival (OS) rate of around 20% was observed[95]. In HCC, the first anti-CTLA-4 tested was tremelimumab, a fully human IgG2 monoclonal antibody. Response rates were more modest in advanced hepatitis C virus-related HCC, with a median OS of 8.2 mo and survival rate of 43% at 1 year[96]. Another study conducted on hepatitis B virus and hepatitis C virus-associated HCC combined tremelimumab with tumor ablation at day 36[97]. Twenty-six percent of patients achieved a partial response with an OS of 12.3 mo. Inversely to melanoma, extensive studies were not conducted in HCC with anti-CTLA-4 antibodies as monotherapies. Ipilimumab is now approved, in combination with the anti-PD-1 nivolumab for previously treated advanced HCC, as detailed below.
The significant results obtained with anti-CTLA-4 therapies are also accompanied with severe adverse events. Dogmas that patients with immune-related adverse events have higher response rates have not been confirmed. Adverse events are mainly immune-related such as rash, thyroiditis and frequent complications of the gastrointestinal tract, including aphthous ulcers, esophagitis, gastritis, diarrhea and colitis in around 20% of patients[98]. These adverse effects could be linked to high expression of CTLA-4 on mucosal Tregs[99]. Liver toxicity with ICI-related hepatitis is also a severe adverse effect of anti-CTLA-4 treatment that could be life-threatening in case of delayed management[100]. Oral glucocorticoids or additional immunosuppressants are usually administered to those patients. After adverse effects, an important question is to restart treatment or not. The decision depends on the severity of the complications and the cancer status[101]. Importantly, retreated patients could develop the same adverse event and others new complications. However, an alternative ICI could be administered to patients with adverse effects,i.e.anti-PD-1 is safety after deleterious ipilimumab treatment in melanoma patients[102].
To limit those toxicities, targeting TILs rather than peripheral populations will be preferred with antibodies against the PD-1/PD-L1 axis, which exhibit less severe adverse events[103]. In addition to fewer immune related adverse events, PD-1/PD-L1 inhibitors also produced greater anticancer activity. Since PD-1 is more broadly expressed than CTLA-4, on tumor cells in particular, and its expression is also induced by chronic antigen exposure, anti-PD-1 antibodies may exert additional anti-tumor effects and exhibits superior clinical activity and safety when compared to anti-CTLA4[104]. The rationale of combining anti-CTLA-4 with anti-PD-1 therapies is also supported by the differential immune patterns observed in individual monotherapies[105].
Another important decision is the selection of anti-PD-1 or anti-PD-L1 therapies. Indeed, PD-L1 inhibition preserves the interaction between PD-1 and its other ligand PD-L2, while it blocks its interactions with CD80, an alternative interaction that has been recently reported to promote T-cell responses[106]. Conversely, PD-1 inhibition blocks the interaction of PD-1 with its two ligands but preserves anti-tumor PDL1/CD80 complexes. Therefore, these antibodies may drive differential anti-tumor immune response. For instance, in non-small-cell lung carcinoma, anti-PD-1 therapies exert better anti-tumor response, while anti-PD-L1 antibodies demonstrate less severe adverse effects[107]. In HCC, three drugs are currently authorized in the United States: The two anti-PD1 nivolumab and pembrolizumab for advanced HCC and one anti-PD-L1, atezolizumab approved in combination with the anti-vascular endothelial growth factor (anti-VEGF) bevacizumab. Nivolumab and pembrolizumab approval has been accelerated by FDA after promising results obtained in preclinical studies on sorafenib refractory HCCs, respectively in Checkmate 040[108] and KEYNOTE-224[109] (20% of overall response rate and 60% of disease control rate). However, in phase 3 trials both agents did not achieve statistical significancy according to the registered statistical plan (CheckMate-459[110] and KEYNOTE-240[111]). New phase 3 trials are conducted for these two drugs as an adjuvant in CheckMate-9DX for nivolumab (NCT03383458), and for pembrolizumab KEYNOTE-937 (NCT03867084) or in secondline with pembrolizumab KEYNOTE-394 (NCT03062358). New anti-PD-1 antibodies are also currently under investigation. The anti-PD-1 tislelizumab, an antibody designed to limit FcγR-mediated phagocytosis, demonstrated a good antitumor activity in a phase 1 trial — a phase 3 trial is ongoing in various solid cancers including non-small cell lung cancer, esophageal squamous cell carcinoma and HCC (RATIONALE 301)[112]. Camrelizumab is also an alternative, which has been tested in China on 220 patients from multiple centers. At a median follow-up at 12.5 mo, the objective response rate (ORR) was 14.7% and 6-mo OS rate was 74.4%. No complete response was observed, 17.6% of patients present partial response and 23.1% a stable disease. The median progression free survival (PFS) was only of 2.6 mo, shorter than other ICIs. Grade 3 and 4 adverse events occurred in 22% of patients[113].
Strategies combining anti-PD-1/PD-L1 with anti-CTLA-4 antibodies have been evaluated in various cancers and in March 2020 FDA have granted approval for nivolumab/ipilimumab (1 and 3 mg/kg) in advanced HCC patients who have priorly received sorafenib. In Checkmate-040, at a median follow-up of 30.7 mo, the combination arm demonstrated 29% ORR. The median duration of response was 21.7 mo. No adverse effects were observed for 79% of patients. An ORR of 31% with 7 complete responses was provided by Blinded independent central review per RECIST[114]. Nonetheless, it has been shown that a combination of ipilimumab and nivolumab leads to higher incidence of ICI-related hepatitis in different cancers including melanoma with 6% to 9% as compared to 1% in single therapies[115]. Rapid diagnosis and management are thus crucial for better outcomes. Another PD-1/CTLA-4 blocking strategy combining durvalumab with tremelimumab is currently under investigation in a randomized, multi-center phase 3 study called HIMALAYA (NCT03298451) to compare combination against durvalumab or sorafenib alone as a first-line therapy for advanced HCC.
Another combination of ICI successfully tested in HCC is atezolizumab plus bevacizumab (anti-VEGF) in first-line in patients with unresectable HCC. A phase III trial (IMbrave150) showed improved progression-free survival of 6.8 movs4.3 mo for sorafenib with an OS at 12 mo of 67.2%vs54.6%[116]. Hypertension, a typical adverse effect of bevacizumab, occurred in 15.2% of patients receiving the combination therapy.
Another intensively tested strategy is to combine ICIs with locoregional treatment, which have demonstrated synergistic activities. Tumor destruction by locoregional treatments releases TAAs promoting immune cell priming, which could be even more enhanced by ICIs. Phase 1, 2 and 3 clinical trials are now conducted with anti-PD-1 or anti-PD-L1, alone or combined with anti-CTLA-4 or anti-angiogenic agents, together with transarterial chemoembolization, hepatic artery infusion chemotherapy or external beam radiation therapy[117] (Table 1). Until now, the combination of ICIs with tyrosine kinase inhibitors such as sorafenib was not concluding. Three phase 3 clinical trials are now conducted to evaluate the benefit of such combinations (NCT04194775, NCT04344158, NCT03755791). However, these recent years, combination of epigenetic drugs with ICIs have emerged as potent therapeutic avenues in hematologic and solid tumors, a point that we will develop in the next paragraph.
EPIGENETICS AND HCC
These recent decades, epigenetic mechanisms have emerged as crucial decisionmakers of cell fate determination and deregulations of epigenetic mechanisms could lead to modifications of gene transcription in the cell, which could favor the initiation and progression of cancers. Conventionally, the epigenetic code is divided into three major mechanisms: ncRNA driven-regulations, DNA methylation and histone modifications mainly occurring on H3 and H4 histones. Many studies have been focusing on miRNA implications in HCC but few data are currently available concerning the clinical used of ncRNA-based therapies in combination with ICIs. We will thus develop the promising results obtained regarding approaches targeting DNA methylation and histone modifiers in HCC, alone or in combination with ICIs (Figure 4).
DNA methylation and DNMT inhibitors
DNA methylation in somatic cells is regulated by DNA methyltransferases that add, in CpG dinucleotide, a CH3group on the 5’ position of the pyrimidine ring in cytosine residue. This modification in methylation will monitor the binding of transcription factors and DNA accessibility in the DNA regulatory region, inevitably leading to modulate gene transcription[118]. The DNMT family is composed of DNMT1, DNMT2, DNMT3A, DNMT3B and DNMT3L. DNMT1 is known to act mainly as a “maintenance” methyltransferase during DNA synthesis and DNMT3A and DNMT3B act as “de novo” methyltransferase during development. But DNMT1 can also act as a “de novo” methyltransferase for genomic DNA and DNMT3A and DNMT3B can also act as “maintenance” methyltransferase during replication[119,120]. The catalytically inactive DNMT3L stimulates the activity of the DNMT3A and DNMT3B enzymes by a direct binding to their respective catalytic domains. Overexpression of DNMTs and their mutations in a variety of tumors, including HCC, modify DNA methylation profiles[121]. Inversely, modification of enzymes involved in DNA demethylation such as TETs (Ten-eleven translocation) is also frequently observed[122]. DNA hypomethylation associated with genome instability and locus-specific hypermethylation of CpG islands are an epigenetic hallmark of cancer, associated with uncontrolled cell proliferation and survival leading to tumor growth. In HCC, DNA methylation is increasingly altered from cirrhosis to preneoplastic lesions and to HCC, without etiology differences, and could be associated with tumor recurrence andsurvival[123-125]. Promoter hypermethylation related to gene silencing is also often observed on tumor-suppressor genes and regulators of cell proliferation and survival such asAPC, CDH1, CDKN1AandCDKN2A[126].

Table 1 Main clinical trials on immunotherapies and epigenetic agents in monotherapies or in combination

Figure 4 Overview of the main epigenetic mechanisms in hepatocellular carcinoma and their inhibitors.
To counteract the tumoral effect of DNA methylation, several DNMT inhibitors (DNMTi) have been extensively studied and under clinical trials for hematologic cancers and increasingly tested in solid tumors. First generation DNMTis like 5-azacytidine (5-aza) and decitabine, can be incorporated into DNA and favor DNMT1 degradation by irreversible binding leading to DNA demethylations. Patients with advanced HCC treated with decitabine show significant clinical benefit from this treatment and a favorable toxicity profile[127]. Second generation DNMTis that are more stablein vivo, have shown interesting results. Zebularine treatment is potentially less toxic, since it does not incorporate into DNA, and gives promising results on an HCC mouse model with high degree of CpG methylation[128]. Guadecitabine was also successfully tested under the clinical trial NCT01752933 on patients which were not responsive to sorafenib with an average PFS of 2.7 mo and an OS of 8 mo[129]. Interestingly, guadecitabine promotes an innate immune response through reactivation of epigenetically silenced endogenous retroviruses and thus could improve ICI sensitivity[130].
HISTONE MODIFICATIONS AND TARGETING DRUGS
Another central epigenetic mechanism is the posttranslational modifications of histones, which control gene expression by modulating chromatin accessibility. Histone-modifying enzymes target specific residues on histone tails by acetylation, phosphorylation or methylation. Other modifications of histone residue exist but are less common, such as ubiquitination, citrullination, ADP-ribosylation, butylation[131]. First, histone acetylation is based on a reversible addition of an acetyl group on histone lysine residues that are added by histone acetyltransferases (HATs) and removed by histone deacetylases (HDACs) (Figure 4). Histone acetylation is often associated with a positive gene transcription. Secondly, like DNA methylation, histone methylation is based on the addition of a methyl group on a lysine or an arginine residue in the histone tails by histone methyl transferases (HMTs). Histone demethylases (HDMs) are responsible for methyl removing. Some histone methylation marks are associated with an active gene transcription, like H3K4me3[132], H3K36me3[133] and H3K79me3[134] and others are rather repressive marks, like H3K27me3[135], H3K20me3[136] and H3K9me3[137]. The expression of several histone modifiers is deregulated in HCC and associated with tumor progression and prognosis, such as HAT with hMOF[138], a plethora of HDAC (HDAC1, 2, 4 and 5, and SIRT1, 2 and 7)[139]. HMT are also concerned with the best characterized EZH2 promoting gene repression through H3K27 trimethylation, G9a[140] and SUV39H1[141] mainly associated with gene repression through H3K9 modifications. Regarding histone modifications, another key actor is BRD4, which reads H3K27ac marks highly enriched in large clusters of enhancers. BRD4 was found overexpressed in HCC and required for super-enhancermediated expression of oncogenes[10].
As DNMTi, HDAC inhibitors (HDACi) have also been evaluated in clinical trials for hematological malignancies but also in solid cancers such as HCC. HDACis bind the zinc-containing catalytic domain of HDACs and thus modify histone acetylation status and gene transcription through HDAC inhibition. An interesting phase 2 clinical study of Yeoet al[142] (NCT00321594) shows the beneficial effect of belinostat in unresectable HCCs. Belinostat, a pan-HAC inhibitor against zinc-dependent HDACs, could increase PFS to 2.6 mo and OS to 6.6 mo with tumor stabilization. The SHELTER study (NCT00943449) combining sorafenib with resminostat, another pan-HDACi targeting HDAC 1, 2 and 3, doubles the OS of advanced HCC patients (8 mo instead of 4.1 mo)[143]. Interestingly, some epigenetics drugs have shown interesting results in HCC experimental studies regarding their impact on tumor microenvironment and tumor response to ICIs. The BET bromodomain inhibitor i-BET762 significantly reduces the level of Monocytic-MDSCs and enhances TILs, alone or in combination with anti-PDL1, and consequently decreases tumor growth in two fibrotic HCC mouse models[144]. In the same way, the co-inhibitor of G9a and DNMT1 called CM-272 favors differentiated HCC and impairs the pro-tumorigenic effects of the surrounding fibrotic stroma[145]. Together, these data support the potent therapeutic benefit of targeting microenvironment remodeling together with epigenetic reprogramming during HCC, in a context of fibrogenesis in particular.
THERAPEUTIC STRATEGIES COMBINING ICI WITH EPIGENETIC DRUGS
Most immunotherapies are based on the targeting of immune checkpoints and the enhancement of immune system reaction to eradicate cancer cells but not all the patients are good responders to those cures. As mentioned previously, several treatments targeting epigenetic mechanisms allow to modify tumor progression and response to treatment. Epigenetic drugs that target DNMTs and HDACs, can in particular upregulate the expression of several immune signaling components in cancer cells such as TAAs[146], stress- and death-induced ligands and receptors, expression of co-stimulatory molecules at the cell surface but also expression of checkpoint ligands[147,148]. Therefore, epigenetic drugs have been used as neoadjuvant agent or in combination with immunotherapies to prime the immune system and create a better response to ICIs.
As previously detailed, cancer cells can evade immune surveillance by a lack of expression of TAAs. Cancer testis antigens (CTAs) are the best characterized TAAs that are regulated by epigenetic events. They are expressed in embryonic and germ cells but silenced by methylation of their promoter in mature somatic and cancer cells. The use of DNA methylation inhibitors such as DNMTis have proved CTAs reexpression in several solid tumors[146,147,149]. HDACis can also induce the reexpression of CTAs but in a less extent than DNMTis, in human cancer cell lines[150]. Several clinical trials are already ongoing (Table 1). Other TAAs are sensitive to several DNMTis or HDACis depending on cancer type and once again DNMTis are more efficient than HDACis[151]. Those drugs can also be used to compensate the methylation deregulation of the promoter region of the APM (antigen processing machinery) component, like TAP-1, TAP-2, LMP-2, LMP-7 and MHC molecule in various tumors[152-154]. Epigenetics drugs can also facilitate tumor cells death by inducing the expression of death receptors, stress induced ligands and co-stimulatory molecules that will sensitize tumor cells to immune-mediated cells lysis[155-161]. Those drugs can also sensitize cancer cells to immune checkpoint therapies targeting PDL-1 and PDL-2, PD-1 and CTLA-4 by increasing their expression on both cancer cells and TILs favorizing their response to ICI[153,154]. Woods and collaborators show on a mouse model of melanoma that a pretreatment with HDACis upregulates PD-L1 and PD-L2 expression and favor the effect of the anti-PD1 treatment, slowing tumor progression and increasing mouse survival[162]. The co-inhibition of H3K27me3 and CTLA-4 reduces the number of Tregs in a mouse model of melanoma and limits tumor size[163]. An interesting work of Goswami and collaborators also shows that the pharmacologic inhibition of EZH2 with CPI-1205 on human T cells altered their Treg phenotype and function and enhanced T cytotoxic activity[164]. They also observe in patients with melanoma or prostate cancer that the anti-CTLA-4 ipilimumab increases EZH2 expression in peripheral T cells. Finally, they could demonstrate in their murine models that EZH2 targeting in T cells could improve the antitumor response mediated by an anti-CTLA-4 therapy. EZH2 appears to be a target of choice since several others works have unveiled its implication in ICI response. Zhouet al[165] also show in an anti-PD1 resistant model of head and neck cancers that EZH2 targeting can restore response to anti-PD1 treatment by increasing antigen specific CD8+ T cell proliferation. Additionally, EZH2 and DNMT1 co-inhibition increases the expression of the Th1 chemokines CXCL9 and CXCL10 in the ID8 ovarian cancer mouse model. This leads to an increase in CD8+ T cell infiltration and improves response to anti-PD-L1 treatment[8]. As previously mentioned, DNMTis also constitute promising partners for ICI, and particularly 5-azacytidine. In a transplantable mammary carcinoma and mesothelioma murine models, the use of 5-azacytidine increases the anti-CTLA-4 antitumor efficiency[166]. A combination of anti-CTLA-4 and anti-PD-1 together with the two epigenetic modulatory drugs 5-azacytidine and the HDACi entinostat could eradicate tumors in mice with colorectal or metastatic breast cancers. These combined strategies mainly inhibit the suppressive activity of Granulocytic-MDSCs against intratumor T cell killing[167]. Many phase 2 trials are currently testing the impact of entinostat with ICI in several cancers (Table 1).
HCC tumors arise in fibrotic livers enriched in MDSCs with less infiltrating lymphocytes inside the tumor[168]. MDSC enrichment is also correlated with an aggressive tumor phenotype and a poor survival rate. Liuet al[144] show on a fibrotic-HCC mouse model that inhibiting monocytic MDSCs with a combination of molibresib, a BET bromodomain inhibitor, with an anti-PD-L1 therapy could enhance TILs and extend mouse survival even with a complete tumor regression[144]. Inhibition of EZH2 and DNMT1 by DZNep and 5-azacytidine respectively, led to tumor regression after anti-PD-L1 treatment of a subcutaneous HCC cell mouse model (HepG2, G-Hep3B and Hepa1-6). This increases cytotoxic T lymphocyte trafficking and promotes cancer cell apoptosis[169]. A second generation of DNMTi molecule, guadecitabine, shows interesting optimization of immunotherapy treatment. Guadecitabine is actually under a clinical trial as a monotherapy in HCC patients and shows a better stability and performance than the first generation DNMTis[130]. Other clinical trials with this DNMTi are actually ongoing in combination with ICI including in HCC (Table 1). HDACi have also been tested in HCC. In a subcutaneous Hepa129 murine model, Llopizet al[170] demonstrate that the HDACi belinostat increases the anti-tumor activity of anti-CTLA-4 therapy. This combination enhances IFN-γ production by T-cells and decreases the number of Tregs. It also induces an early upregulation of PD-L1 on tumor-specific APCs and delay PD-1 expression on TILs. Furthermore, belinostat combined to CTLA-4 and PD-1 blockade leads to a complete tumor rejection[170].
CONCLUSION
The liver is a highly complex organ which orchestrates fundamental metabolisms finely regulated at the transcriptional and epigenetic level. Liver parenchyma also harbors a specialized immune system playing a central role in liver homeostasis with the constant management of toxins, diet or bacteria susceptible to trigger deleterious inflammation. However, when toxin and pathogenic insults get into chronicity, liver inflammation could sensitize to cancer development in part by immune suppression mechanisms. Thus, this peculiar tumor microenvironment constitutes an interesting opportunity to therapeutic avenues based on ICIs. Due to its high complexity, HCC response to conventional therapies is quite heterogeneous and frequently associated with poor outcome, rendering this cancer one of the deadliest cancers in the world. While several solid tumors are good responders to immunotherapy, ICIs in HCC show disappointing results, especially on β-catenin mutated HCCs, even if ICIs have given better results than tyrosine kinase inhibitors particularly in terms of prolonged response. Contrary to other solid tumors, personalized therapies for HCC are more complex to define, in particular because of tumor appearance in a context of cirrhotic livers with high level of inflammation and damages. Even if genomic analyses of the tumor mutational background have already classified HCCs, a translational approach taking into account the immune cell pattern, inside and outside the tumor, but also their respective epigenetic state, regarding DNA methylation level or histone marks, will be of therapeutic benefit to select the more efficient therapy for each patient. The bi-therapy combining immunotherapies either with anti-angiogenic agents or epigenetic drugs currently appears as the most promising to treat HCC patients. It is now well known that multiple epigenetic modulations can lead to the modification of tumor microenvironment by expressing TAAs, immune checkpoint ligands, costimulatory molecules and death-induced ligands or receptors at the cell surface. Therefore, using epigenetic agents to prime the microenvironment before immunotherapy may favor a better outcome for patients with a re-polarization of immune cells towards an efficient anti-cancer response. Several clinical studies have already shown that these bi-therapies are efficient in different solid tumors like pulmonary cancer, melanoma and colon cancers. Recently, results from clinical trials with epigenetic drugs and immunotherapy on advanced HCC patients showed interesting results with an extension of patient OS. These new combined therapies could be the new hope for HCC treatment. However, these clinical trials were only performed on advanced HCCs and it would be necessary to test these on HCC of lower grade because these treatments may be more efficient on these subgroups. The important point in close future is to identify predictive biomarkers, based on patient responses during clinical trials, to predict patient that will respond to treatment or not. Correlative studies are thus a prerequisite to create guidelines for personalized treatments and sequencing therapies to counteract immune dysfunction and overcome the current barriers to immunotherapies in HCC.
杂志排行

World Journal of Hepatology的其它文章
- Coronavirus disease 2019 and non-alcoholic fatty liver disease
- Advances in the management of cholangiocarcinoma
- Herbal and dietary supplement induced liver injury: Highlights from the recent literature
- Challenges in the discontinuation of chronic hepatitis B antiviral agents
- Liver Kidney Crosstalk: Hepatorenal Syndrome
- Hepatitis C virus treatment failure: Clinical utility for testing resistance-associated substitutions