伴SCN1A基因突变Dravet综合征的临床电生理特点
2021-10-09徐那杨莉李玉芬徐丽云邱世彦孙学梅
徐那 杨莉 李玉芬 徐丽云 邱世彦 孙学梅
[摘要] 目的 探讨伴SCN1A基因突变Dravet综合征的临床电生理特点,为该疾病的临床诊断提供依据。方法 回顾性分析2014年6月—2018年1月临沂市人民医院收治的11例伴SCN1A基因突变Dravet综合征病儿的临床资料,包括智力发育、颅脑影像学、视频脑电图(背景活动、痫样放电、发作期图形)等。结果 11例病儿,男5例,女6例;年龄1岁5个月~4岁,中位起病年龄5个月(2~11个月),确诊年龄9个月~3岁6个月。6例以复杂性热性惊厥起病,5例以无热局灶性抽搐发作起病。1岁之前有发热诱发癫痫发作持续时间大于15 min者3例,大于30 min者7例;24 h内出现热性惊厥≥2次者8例;1岁前出现无热惊厥10例;11例病儿均曾出现半侧阵挛或局灶性发作。癫痫发作类型均为强直阵挛或局灶性发作,最长无发作时间1~12个月。11例病儿起病前发育均正常,起病后有不同程度落后。颅脑MRI均未见明显异常。11例病儿脑电图背景慢化不明显,2例发作间期有局灶性痫样放电,1例为广泛性棘慢波,8例为正常或睡眠期双导小棘波,3例异常者复查结果为正常或睡眠期雙导小棘波。应用多重连接依赖性探针扩增(MLPA)技术检测SCN1A基因,11例病儿均检测到新生突变,其中大片段缺失1例,无义突变2例,错义突变8例。结论 伴SCN1A基因突变Dravet综合征多以复杂性热性惊厥起病,易出现癫痫持续状态,起病早期脑电图及颅脑MRI多正常,可不出现肌阵挛等发作类型,当临床遇到类似病儿,基因检测有助于早期确诊。
[关键词] 癫痫,肌阵挛性;NAV1.1电压门控钠通道;突变;脑电描记术
[中图分类号] R742.1
[文献标志码] A
[文章编号] 2096-5532(2021)04-0527-05
Dravet综合征是婴儿期起病的由遗传因素引起的癫痫性脑病,为一种常染色体显性遗传病,其总体发病率为1/40 000~1/20 000[1-3],男女比例约为2∶1。既往研究表明,Dravet综合征的主要临床特点为婴儿期起病,起病前生长发育正常,起病初表现为全面性、单侧性或单侧交替性的热性或非热性惊厥,早期脑电图、颅脑磁共振检查阳性率低,起病后可出现肌阵挛、不典型失神等多种类型发作,同时伴有发育落后、共济失调等症状,对各类型抗癫痫药物治疗反应差[4-6]。由于病情严重、表型复杂,Dravet综合征早期往往被误诊,而在疾病早期不规范的抗癫痫治疗可对病儿的认知结果产生负面影响,故早期诊断是关键[7]。研究已证实,该综合征超过80%是由电压门控钠通道基因SCN1A突变所致[8-9]。SCN1A基因突变是癫痫对大脑产生长期有害影响的先决条件,表明癫痫发作与基因突变之间存在明显的相互作用,从而形成由病理重塑产生的严重表型[10-11]。本研究对我院收治的11例伴SCN1A基因突变Dravet综合征病儿的临床电生理特点进行总结分析,旨在探讨如何尽早明确诊断,避免误诊误治,为临床遗传咨询及早诊断、早治疗提供依据,从而改善预后。现将结果报告如下。
1 对象与方法
1.1 研究对象
2014年6月—2018年1月,选择临沂市人民医院收治的11例伴SCN1A基因突变Dravet综合征病儿为研究对象。其临床诊断标准如下:①1岁以内常以热性惊厥起病(高峰年龄为生后6个月),多表现为长时间的全面性或半侧阵挛发作;②1岁后可出现肌阵挛发作、不典型失神发作、局灶性发作等多种发作类型;③惊厥具有热敏感;④易发生惊厥持续状态;⑤早期发育正常,1岁以后逐渐出现精神运动发育落后或倒退现象,可有共济失调和锥体束征;⑥脑电图在1岁以前多为正常,1岁以后出现全导棘慢波、多棘慢波或局灶性、多灶性痫样放电;⑦抗癫痫药物疗效差;⑧可有热性惊厥史或者癫痫家族史[4-5,12];⑨根据美国医学遗传学和基因组学学院指南(ACMG)评估SCN1A基因突变的致病性,结合临床确定病儿的致病基因。
1.2 研究方法
1.2.1 病史资料采集 采集11例Dravet综合征病儿的起病年龄、确诊年龄、癫痫发作类型、生长发育史、既往疾病史、家族史、智力发育、颅脑影像学、视频脑电图(背景活动、痫样放电、发作期图形)、治疗过程等临床资料。
1.2.2 基因检测 采集病儿及其双亲的外周血各2 mL,置于含乙二胺四乙酸的抗凝试管中。采用二代高通量目标区域准确捕获测序进行癫痫基因测定,再利用人类基因突变数据库和千人基因组数据库等相关数据库分析数据,借助蛋白损伤相关预测软件预测突变,探索与疾病有关的可疑致病性以及致病性基因突变。采用Sanger测序法进行验证分析,对缺乏明确致病性基因突变病儿,通过多重连接依赖性探针扩增(MLPA)技术检测SCN1A基因的大片段变异。
2 结 果
2.1 临床特点
本组11例病儿,男5例,女6例;年龄1岁5个月~4岁;起病年龄2~11个月,中位起病年龄5个月;确诊年龄9个月~3岁6个月。6例以复杂性热性惊厥起病,余5例以无热局灶性发作起病。1岁前曾有发热诱发癫痫发作持续时间大于15 min者3例,大于30 min者7例;24 h内出现热性惊厥≥2次者8例;1岁前出现无热惊厥10例;11例病儿均曾出现半侧阵挛和(或)部分性发作。3例有时发热无抽搐发作,其余8例逢热必抽。癫痫发作类型均为强直阵挛或部分性发作,未出现肌阵挛及其他发作类型,最长无发作时间1~12个月。11例病儿起病前发育均正常,起病后发育有不同程度落后。
2.2 影像学及视频脑电图特点
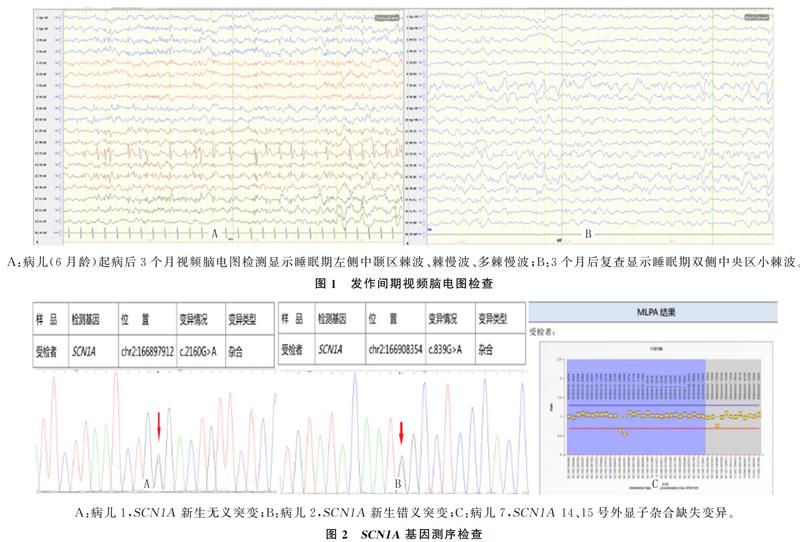
本组病儿颅脑MRI均未见明显异常。11例病儿脑电图背景慢化均不明显,2例发作间期有局灶性痫样放电,1例为广泛性棘慢波,8例为正常或睡眠期双导小棘波,3例异常者复查结果为正常或睡眠期双导小棘波。见图1。
2.3 分子遗传学检查
本研究11例病儿SCN1A基因均阳性,1例MLPA检测到大片段缺失,2例為无义突变,8例为错义突变,11例均为新生突变,突变位点分别为c.2160G>A、c.5531C>A、c.1076A>G、c.4502C>A、c.755T>A、c.1129C>T、c.677C>G和c.839G>A。见图2。
3 讨 论
1978年,DRAVET等首次报道了Dravet综合征,该病以前称为婴幼儿严重肌阵挛性癫痫,2001年国际抗癫痫联盟推荐名为Dravet综合征,为常染色体显性遗传病。Dravet综合征是一种难治性癫痫综合征,多在婴儿期起病,早期常表现为热性惊厥,1岁后出现多种形式的无热惊厥,由于其病情严重、表型复杂,早期往往被误诊,错误的诊断又导致错误的治疗,从而导致医源性病情加重,如使用钠通道阻滞剂类的抗癫痫药物则会导致发作增加[12]。有学者研究发现,Dravet综合征病儿发生癫痫性猝死的危险性高于其他癫痫病儿,而癫痫持续状态后发生急性脑病是Dravet综合征病人早期死亡的原因[13-16]。本研究拟总结Dravet综合征病儿的临床电生理本特点,以期为该病的临床早诊断、早治疗提供依据,降低误诊率,改善预后。结合既往文献以及本研究,总结Dravet综合征病儿的临床特点如下。①有癫痫及热性惊厥家族史。②1岁以内起病,6个月为起病高峰期,起病前发育正常。③疾病初期多表现为局灶性发作或强直阵挛发作,后期可出现各种发作表现,如肌阵挛发作、不典型失神发作以及全面性、单侧或双侧交替的热性或非热性惊厥及癫痫持续状态(也可不出现,如本研究11例病儿均未出现除局灶性发作及强直阵挛发作以外的其他癫痫发作类型);癫痫发作的频率和严重程度随发育成熟而减少[17-18]。④病初脑电图正常,后期出现局灶、多灶性或广泛性放电。本组病儿如未出现除局灶性发作及强直阵挛发作之外的其他发作类型时脑电图可不恶化,脑电图背景亦可为正常,即使起病早期发作频繁时发作间期出现一过性癫痫样放电,如果发作不加重,脑电图可再次恢复正常。随着年龄增长可能会出现年龄相关性放电,如Rolandic区或枕区等局灶性放电及广泛性棘慢波,放电的出现并不加重发作程度,所以临床上遇到此类情况可以先不用调整抗癫痫药,而既往文献对此研究较少。⑤随病程进展出现精神运动发育迟缓或倒退以及认知功能障碍。⑥抗癫痫药物治疗效果差,尤其是奥卡西平及拉莫三嗪等可能加重发作。⑦多数有SCN1A基因相关突变,突变类型大部分为点突变,如错义突变、无义突变、移码突变和剪切位点突变等,少数可以为片段缺失或重复[19-20]。本组中10例为点突变,1例为片段缺失。因此,对于二代测序未发现SCN1A基因突变的病儿,应常规进行SCN1A基因MLPA筛查,以发现基因内部的片段缺失或者重复。但是,Dravet综合征也有一些其他的致病基因被报道,如PCDH19、SCN2A、SCN8A、SCN9A、SCN1B、PCDH19、GABRA1、GABRG2、STXBP1、HCN1、CHD2和KCNA2等[18,21],故对于临床表型符合Dravet综合征的病儿,若SCN1A基因Sanger测序和MLPA检测均为阴性,应进一步筛查其他致病突变,为明确病因提供线索。
目前,Dravet综合征的治疗除了控制癫痫发作外,治疗其并发症、共患病也很重要,因为它们对Dravet综合征病儿的生活质量有很大影响。本病治疗困难,通常无法达到完全的癫痫发作控制[22],目前尚没有长期有效的治疗方案。癫痫发作通常可以被钠通道阻滞剂类抗癫痫药物加重。推荐的一线药物包括氯巴占和丙戊酸,但这些药物很难完全控制癫痫发作,托吡酯、左乙拉西坦、生酮饮食[23-24]和迷走神经刺激[25-26]可以选择作为辅助治疗。目前正在开发的几种药物,特别是芬氟拉明[27]和大麻二酚[28-29],在临床试验中显示出功效。亦有文献报道,雌孕激素类是γ-氨基丁酸A受体的正调节剂,可减少癫痫发作或终止癫痫持续状态[29-31]。Dravet综合征病人易出现癫痫持续状态,特别是在儿童早期,故所有病人都应积极行家庭内抢救治疗(通常用苯二氮类药物),如果癫痫持续存在,应尽快到医院行进一步治疗。
综上所述,Dravet综合征病儿起病年龄早,多数在1岁内以无热局灶性发作或复杂性热性惊厥起病,1岁前多出现癫痫持续状态,对热敏感;起病早期脑电图及颅脑MRI多正常;起病前发育均正常,起病后可有轻中度的发育落后。当临床遇到此类病儿时,应高度怀疑Dravet综合征的可能,尽早进行SCN1A基因点突变及MLPA筛查,避免误诊、误治,早诊断、早治疗可以有效改善预后。当Dravet综合征病儿脑电图由正常转为异常放电时,应根据临床发作调整药物,而不是单纯依据视频脑电图结果的变化,避免临床过度治疗。
[参考文献]
[1]BAYAT A, HJALGRIM H, MLLER R S. The incidence of SCN1A-related Dravet syndrome in Denmark is 1∶22, 000: a population-based study from 2004 to 2009[J]. Epilepsia, 2015,56(4):e36-e39.
[2]BRUNKLAUS A, ELLIS R, STEWART H, et al. Homozygous mutations in the SCN1A gene associated with genetic epilepsy with febrile seizures plus and Dravet syndrome in 2 families[J]. European Journal of Paediatric Neurology, 2015,19(4):484-488.
[3]GIL-NAGEL A, SANCHEZ-CARPINTERO R, SAN ANTONIO V, et al. Ascertaining the epidemiology, patient flow and disease management for Dravet syndrome in Spain[J]. Rev Neurol, 2019,68(2):75-81.
[4]XU X J, ZHANG Y H, SUN H H, et al. Early clinical features and diagnosis of Dravet syndrome in 138 Chinese patients with SCN1A mutations[J]. Brain and Development, 2014,36(8):676-681.
[5]BATTAGLIA D, RICCI D, CHIEFFO D, et al. Outlining a core neuropsychological phenotype for Dravet syndrome[J]. Epilepsy Research, 2016,120:91-97.
[6]HE N, LI B M, LI Z X, et al. Few individuals with Lennox-Gastaut syndrome have autism spectrum disorder: a comparison with Dravet syndrome[J]. Journal of Neurodevelopmental Disorders, 2018,10(1):10.
[7]DE LANGE I M, GUNNING B, SONSMA A C M, et al. Influence of contraindicated medication use on cognitive outcome in Dravet syndrome and age at first afebrile seizure as a clinical predictor in SCN1A-related seizure phenotypes[J]. Epilepsia, 2018,59(6):1154-1165.
[8]DO T T H, VU D M, HUYNH T T K, et al. SCN1A gene mutation and adaptive functioning in 18 Vietnamese children with dravet syndrome[J]. Journal of Clinical Neurology, 2017,13(1):62-70.
[9]JIANG T J, SHEN Y P, CHEN H, et al. Clinical and molecular analysis of epilepsy-related genes in patients with Dravet syndrome[J]. Medicine, 2018,97(50):e13565.
[10]SALGUEIRO-PEREIRA A R, DUPRAT F, POUSINHA P A, et al. A two-hit story: seizures and genetic mutation inte-raction sets phenotype severity in SCN1A epilepsies[J]. Neurobiology of Disease, 2019,125:31-44.
[11]RITTER-MAKINSON S, CLEMENTE-PEREZ A, HIGA-SHIKUBO B, et al. Augmented reticular thalamic bursting and seizures in Scn1a-Dravet Syndrome[J]. Cell Rep, 2019,26(1):54-64.e6.
[12]曾琦,張月华,杨小玲,等. Dravet综合征患儿SCN1A基因片段缺失及重复的研究[J]. 中华医学遗传学杂志, 2017,34(6):787-791.
[13]TIAN X J, YE J T, ZENG Q, et al. The clinical outcome and neuroimaging of acute encephalopathy after status epilepticus in Dravet syndrome[J]. Developmental Medicine & Child Neurology, 2018,60(6):566-573.
[14]DRAVET C. Acute encephalopathy after febrile status epilep-ticus: an underdiagnosed, misunderstood complication of Dravet syndrome[J]. Developmental Medicine & Child Neurology, 2018,60(6):534.
[15]MYERS K A, MCMAHON J M, MANDELSTAM S A, et al. Fatal cerebral edema with status epilepticus in children with Dravet syndrome: report of 5 cases[J]. Pediatrics, 2017,139(4):e20161933.
[16]MYERS K A, SHEVELL M I, SBIRE G. Sudden unexpec-ted death in GEFS+ families with sodium channel pathogenic variants[J]. Epilepsy Research, 2019,150:66-69.
[17]GATAULLINA S, DULAC O. From genotype to phenotype in Dravet disease[J]. Seizure, 2017,44:58-64.
[18]STEEL D, SYMONDS J D, ZUBERI S M, et al. Dravet syndrome and its mimics: beyond SCN1A[J]. Epilepsia, 2017,58(11):1807-1816.
[19]SUN H H, ZHANG Y H, LIU X Y, et al. Analysis of SCN1A mutation and parental origin in patients with Dravet syndrome[J]. Journal of Human Genetics, 2010,55(7):421-427.
[20]SHI X Y, WANG J W, KURAHASHI H, et al. On the likelihood of SCN1A microdeletions or duplications in Dravet syndrome with missense mutation[J]. Brain and Development, 2012,34(8):617-619.
[21]CHEMALY N, LOSITO E, PINARD J M, et al. Early and long-term electroclinical features of patients with epilepsy and PCDH19 mutation[J]. Epileptic Disorders, 2018,20(6):457-467.
[22]KNUPP K G, WIRRELL E C. Treatment strategies for Dravet syndrome[J]. CNS Drugs, 2018,32(4):335-350.
[23]DRESSLER A, TRIMMEL-SCHWAHOFER P, REITHO-FER E, et al. Efficacy and tolerability of the ketogenic diet in Dravet syndrome-Comparison with various standard antiepileptic drug regimen[J]. Epilepsy Research, 2015,109:81-89.
[24]LIU F Y, PENG J, ZHU C H, et al. Efficacy of the ketogenic diet in Chinese children with Dravet syndrome: a focus on neuropsychological development[J]. Epilepsy & Behavior, 2019,92:98-102.
[25]DIBU-ADJEI M, FISCHER I, STEIGER H J, et al. Efficacy of adjunctive vagus nerve stimulation in patients with Dravet syndrome: a meta-analysis of 68 patients[J]. Seizure, 2017,50:147-152.
[26]ALI R, ELSAYED M, KAUR M, et al. Use of social media to assess the effectiveness of vagal nerve stimulation in Dravet syndrome: a caregiver's perspective[J]. Journal of the Neurological Sciences, 2017,375:146-149.
[27]SCHOONJANS A, PAELINCK B P, MARCHAU F, et al. Low-dose fenfluramine significantly reduces seizure frequency in Dravet syndrome: a prospective study of a new cohort of patients[J]. European Journal of Neurology, 2017,24(2):309-314.
[28]DEVINSKY O, PATEL A D, THIELE E A, et al. Rando-mized, dose-ranging safety trial of cannabidiol in Dravet syndrome[J]. Neurology, 2018,90(14):e1204-e1211.
[29]GROTHE S, KLUGER G, LOTTE J. Seizure freedom in patients with Dravet syndrome with contraceptives: a case report with two patients[J]. Neuropediatrics, 2018,49(4):276-278.
[30]SAPORITO M S, GRUNER J A, DICAMILLO A, et al. Intravenously administered ganaxolone blocks diazepam-resistant lithium-pilocarpine-induced status epilepticus in rats: comparison with allopregnanolone[J]. Journal of Pharmacology and Experimental Therapeutics, 2019,368(3):326-337.
[31]AMENGUAL-GUAL M, SNCHEZ FERNNDEZ I, WAI-NWRIGHT M S. Novel drugs and early polypharmacotherapy in status epilepticus[J]. Seizure, 2019,68:79-88.
(本文編辑 马伟平)