1例先天性结直肠肛门发育缺陷性疾病
——肛门畸形合并巨结肠病例报告及文献回顾
2021-09-30刘访苏丹张恒鲜振宇任东林
刘访,苏丹,张恒,鲜振宇,任东林
中山大学附属第六医院肛肠外科 广东广州 510655
先天性肛门直肠畸形(anorectal malformation,ARM)是一种复杂的先天性肛门、直肠和泌尿生殖系统疾病,位居新生儿消化道畸形之首,其发病率为1/5 000~1/1 500,但同时合并先天性巨结肠(Hirschsprung’s disease,HSCR)却十分少见,有学者曾报道其发病率为1/250 000~1/75 000[1-3]。HSCR是由于某一肠段的肠肌间神经丛神经节细胞缺如,导致该神经所支配肠段的蠕动收缩功能丧失,病变肠段近端结肠由于肠内容无法排出而被动继发性扩大,故又称先天性无神经节细胞症,是小儿外科较常见的先天性消化道畸形。HSCR发病率为1/5 000~1/2 000,仅次于肛门直肠畸形,居先天性消化道畸形第二位[4],男女比例为(3~4):1,且其有遗传倾向[5]。
ARM合并HSCR是一种较为罕见的先天性消化道畸形,其确切发生率尚不清楚,据文献报道,ARM合并HSCR在消化道畸形患者中的比例为2.3%~3.4%[6-8]。目前关于ARM合并HSCR的文献报道相对较少,且多为单中心、单发病例。近年来,尽管对ARM合并HSCR的诊断有了新的认识,但仍缺乏相应的临床诊疗指南,在手术方式的选择及术后生活质量管理等方面也缺乏充足的经验。
本文通过对1例ARM合并HSCR的成人病例的发病特点、诊疗思路、手术策略及术后生活质量管理等方面进行详细总结,以期提高广大同行对该类疾病的认识,为临床诊疗提供参考。
1 临床资料
1.1 病史及查体
患者男性,29岁,因“排粪困难、异常29年”就诊。自诉出生后诊断为先天性肛门闭锁(无肛门),于出生后第2天行肛门成形术,具体术式不详。术后至今一直粪便干结、排出困难,同时伴有控便能力下降,表现为粪便从肛门自行溢出,次数不规律。无恶心、呕吐、腹痛、腹胀、里急后重感,无肛门流血、流液。为寻求进一步系统诊治遂至我院就诊,2021年4月19日门诊以“便秘、先天性肛门畸形”收入院。
既往史:既往体健,否认高血压病、冠心病、糖尿病等慢性病史,否认肝炎、伤寒、结核等传染病史,否认外伤、其他手术史,否认输血史,否认药物、食物过敏史,预防接种史不详。
个人史:原籍(宁夏)出生长大,无外地居住史,无疫区居住史,无疫水、疫源接触史。无吸烟史,无嗜酒史,无冶游史,无放射性物质、毒物接触史。
婚育史:未婚未育。
家族史:家族中无相关疾病记载,否认患传染病及遗传病等病史。
入院查体:生命征平稳。专科检查:患者取左侧卧位,肛门未见溢液,肛周皮肤无皮疹、糜烂,肛门外观畸形,洞状肛门,见既往手术瘢痕。直肠指诊:肛门口瘢痕性狭窄,小指尖无法通过。
1.2 实验室及辅助检查
血常规、生化全套、凝血功能四项、粪便常规+潜血、术前八项及心电图、胸部CT均未见异常。
术前钡灌肠造影X线检查提示:近端直肠及乙状结肠明显扩张、结肠袋消失、最宽处约19.5 cm,肠腔内见大量粪便堆积(见图1)。

图1 术前钡灌肠造影 X线检查所见
术前肛管MRI检查提示:肛管结构发育不完全,仅有左侧少许肛提肌显示,肛门内外括约肌显像缺失,原肛门被瘢痕组织代替,肛管直肠环结构缺失(见图2)。

图2 术前肛管MRI检查结果
其他检查:因肛管狭窄无法行电子结肠镜、肛管直肠测压等检查。
2 治疗方案
2.1 手术方式
该病例于2021年4月29日行手术治疗,在手术方式选择方面遵循个体化原则:患者为先天性肛门闭锁伴重度肛门括约肌发育不良,行肛门成形术后出现肛管瘢痕性狭窄,在行钡灌肠造影X线检查时发现近端直肠及乙状结肠明显扩张。尽管因患者肛管狭窄无法行结肠镜、肛门直肠测压及病理检查,术前无法明确先天性巨结肠诊断,但近端直肠及乙状结肠肠管因长期扩张而丧失正常功能可能性大,因此考虑采用剖腹探查和远端扩张结肠切除的治疗策略。结合查体和肛管MRI检查结果,患者肛管瘢痕明显,肛门括约肌复合体几乎完全缺失,且扩张肠管向远端累及至肛门口,提示神经节缺失结肠段位于超低位直肠可能性大。因此,如采用一期肠管低位吻合的方式,不仅增加术后吻合口漏的发生风险,还可能导致病变肠段残留、不能完整切除。此外,因肛管功能性结构的先天性缺失,术后重建的肛门“完全无功能”的可能性极大。这种方式无法让患者从经济、安全和生活质量方面得到改善。为达到患者获益最大化,术前即放弃行一期肠管吻合术而改行永久性结肠造口术。首先,采用剖腹探查的方式评估扩张肠管近端累及降结肠中段,适当刺激结肠脾曲远端5 cm左右形态正常的肠管具有蠕动收缩功能,遂决定行远端结肠、直肠切除,降结肠造口术。远端直肠游离至盆膈出口瘢痕处,未行肛门切除术。术中情况见图3。

图3 术中情况
2.2 术后处理
术后予以预防感染、补液、镇痛、营养支持等治疗。
2.3 术后病理结果
术后病理结果提示:巨结肠。结肠肠壁组织,肠壁四层结构清晰,表面上皮脱落,黏膜下层血管扩张、充血,血管壁增厚,间质纤维组织增生,固有肌层浅层增厚,局部平滑肌细胞增大,胞浆淡染,呈空泡样变性,核增大,呈圆形或卵圆形,深肌层局部稍变薄,狭窄段肠管肌间、黏膜未见明显神经节细胞,局部区域纤维组织增生,符合先天性巨结肠病理改变(见图4)。
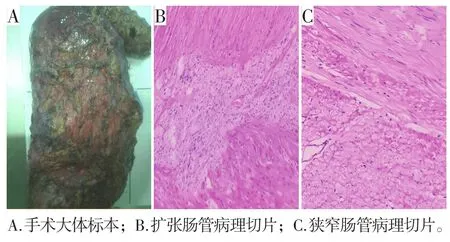
图4 病理标本及病理结果(B、C苏木精—伊红染色,×20)
2.4 术后随访
术后1个月电话随访,患者诉伤口恢复良好,无腹胀、腹痛,无特殊不适,造口排粪正常,饮食质量明显改善,生活学习不受影响。通过电话问答方式采用欧洲癌症研究与治疗组织(European organi⁃sation for research and treatment of cancer,EORTC)结直肠癌生活质量问卷(QLQ-CR 29)评估患者生活质量,该量表由6个维度(排尿问题、腹部和盆腔疼痛问题、排粪问题、大便失禁、焦虑、身体形象)共11个条目组成,该次评估结果显示患者对手术及术后伤口恢复较为满意,但在社会活动、心理状态方面得分较低,医护人员根据评估结果对患者进行沟通和指导,拟后续定期随访观察。
3 讨论
人体结直肠肛门的发育始于胚胎时期第3周,该时期内胚层出现原始消化管,分前、中、后肠,后肠则衍化成横结肠后半、降结肠、乙状结肠、直肠和肛管上段。至第8周,原始肛门部出现凹陷(原始肛穴)并不断向头侧发展,逐渐接近直肠,二者接触后,肛膜破裂,形成肛门并与直肠相通。而胚胎时期肠神经系统发育由起源于神经管的神经嵴,由头端向尾端逐渐发育完成,至第12周完成从食管到肛管整个消化道的移行过程。
先天性肛门直肠畸形,又称肛门闭锁,为小儿最常见消化道畸形。肛门直肠畸形,往往伴发其他畸形,发生率为28%~72%,伴发畸形最多的为泌尿生殖系统畸形,其次为脊柱(特别是骶椎畸形),再次为消化道、心脏以及其他各种畸形。肛门直肠畸形的病理改变很复杂,不仅肛门直肠本身发生发育缺陷,同时盆底肌肉、骶骨、神经及肛周皮肤等均有不同程度的病理改变,肛门直肠畸形的位置越高,这种改变越明显、越严重。因此,先天性肛门直肠畸形的手术难度和术后生活质量随着肛门闭锁的位置和伴随的其他发育异常的不同而不同。
先天性巨结肠是一种常见的消化道疾病,多见于男性,该病是由于远端结肠神经节细胞缺如导致病变肠管痉挛狭窄继而引起近端肠管扩大增粗肥厚和功能性肠梗阻。但该病的病因是远端结肠无神经节细胞,因此更准确的病名为“肠管无神经节细胞症”或HSCR。解剖学家Frederick Ruysch最早(1691年)发现HSCR[9]。HSCR的病理改变为远端结肠或直肠无神经节细胞存在,以致丧失蠕动收缩功能,发生痉挛狭窄[10]。HSCR的诊断及治疗比较复杂,其术前的检查、诊断及手术方法的选择,术后并发症的处理等仍面临困难及挑战[11]。但近年来有研究报道,部分肠管疾病并不是由肠神经节细胞缺如引起,而是由于肠神经节细胞减少、发育不良、未成熟等引起,统称为巨结肠同源病(allied diseases of Hirschsprung’s dis⁃ease,HAD)。由于HSCR与HAD的病理改变、治疗及预后等方面存在较大的差异,所以术前鉴别诊断非常重要。HAD于1958年由Ravlth首先提出[12],这类疾病的症状与HSCR酷似,但病变肠管有神经节细胞存在,治疗可尝试保守治疗,手术方式也与HSCR不同,手术方式不恰当则极易复发。
HSCR患者的临床表现因病变肠管长度的不同而不同,典型的临床表现是在新生儿期出现胎粪排出延迟及腹胀[1,13]。对于先天性肛门直肠畸形患者,如术后出现便秘持续并加重且保守治疗无效时,应警惕合并先天性巨结肠的可能。Oh等[14]研究发现,有50%~60%的ARM患者至少同时有另一种类型的器官发育异常,最常见的为泌尿生殖系统、心血管系统和脊柱/脊椎异常,ARM泄殖腔型发病率最高(93.9%)。ARM合并HSCR时,由于肛门直肠畸形为患者的突出表现,术后出现便秘症状时,临床医师容易混淆诊断而无法明确是否合并HSCR,往往导致HSCR的漏诊,以至于有的HSCR患者到成年才被发现。成人HSCR的诊断难点在于合并既往肠道切除手术史的患者,其在肠切除术后容易发生肠粘连及肠扭转,加上年轻医师对这一疾病认识不足,往往不易联想到合并巨结肠的可能,导致漏诊。
本例患者结合病史、辅助检查、术中情况和术后病理,确诊成人HSCR。对于合并先天性肛门闭锁的患儿,很难同时在术前完善病理检查。对于肛门成形术后以“排粪困难”或“控便功能下降”为主诉的患儿或成人,就诊时必须完善系统检查,评估靠近肛管的肠道形态、重建肛管的结构与功能以及肛管周围肌肉的发育情况。
目前ARM合并HSCR的诊断对临床医师仍是一种挑战,往往需要联合多种检查方法协助诊断,临床上常用的检查方法有肛管直肠测压(anorectal ma⁃nometry,ARM)、钡灌肠(barium enema,BE)、直肠活检(rectal biopsy,RB)等,其中RB是诊断HSCR的金标准[15],准确地说,良好的取材和病理专家的准确判断才是RB诊断HSCR的金标准[16]。直肠活检在西方国家开展较为普遍,欧洲小儿外科协会的成员均认可通过RB确诊HSCR[17-18]。临床上ARM是否合并HSCR可以通过钡灌肠影像学检查及直肠壁组织学检查加以鉴别,但有部分患者在术前不易取到肠壁组织进行病理检查,此时,我们有必要借助其他辅助检查以指导手术治疗,如本例患者中,我们将钡灌肠造影X线检查与肛管MRI结合,可发现肛门直肠的发育缺陷,进一步明确其病灶情况,对制定手术策略具有十分重要的意义。
对HSCR的治疗策略是临床医师最为关注的重点之一,在HSCR手术治疗中,手术原则是切除无神经节细胞肠管等病变肠段并且重建肠道功能。对于诊断明确的HSCR患者,在恰当的手术时机选择合适的手术方式,减少手术相关并发症和再次手术的可能,提高患者生活质量,是我们每一位肛肠外科医师的追求。在HSCR术式方面,腹会阴联合拖出式直肠乙状结肠切除术(Swenson术)开创了先河,此后诸多术式在此基础上加以改进,如Duhamel术、Soave术、Rehbein术等已成为HSCR根治术的主流术式,但各有利弊。关于手术方式优劣的研究目前以回顾性研究和Meta分析为主,尚缺乏高质量证据的研究[19-21]。大多数的HSCR根治术后的排粪异常,如先天性巨结肠相关性小肠结肠炎(Hirschsprung’s associated enterocolitis,HAEC)、便秘复发、肛门有粪便溢出等均可以通过保守治疗治愈,但仍有部分患者(1%~10%)需要再次手术治疗[22]。HSCR根治术后发生便秘的概率为11%~42%[23],首要原因是未能彻底切除病变肠管,导致无神经节细胞病变肠管或神经节细胞发育异常肠管(狭窄段/移行段)残留[24];其次是由于各种原因导致术后末端直肠缺血产生的继发性神经节细胞凋亡或变性[25]。手术成功与否取决于术者对直肠肛管解剖学与生理学的透彻理解及临床诊疗经验积累。HSCR手术方案的选择要求个体化,即不仅取决于患者情况,更取决于医师对何种手术方式更加擅长,以及医疗设备能满足手术操作的需要。一个优秀的肛肠外科医师应当掌握HSCR的各种术式,对不同病情的患者采用不同手术方案,但对于常规病例,鉴于目前没有足够证据表明何种术式最优,术者宜专注于最擅长的术式,掌握所需的技术细节,以最大程度减少并发症的发生。因此,我们在制定诊疗方案时需以患者为中心,让患者及家属参与进来,详细告知诊疗策略及各方案的优缺点,权衡利弊,实现患者获益最大化。
术后的关注重点是患者的生活质量,但生活质量较难准确衡量,受主客观因素的影响。生活质量已成为慢性疾病治疗效果评价的重要终点指标,在评价HSCR患者生活质量的有关研究时,遇到的主要问题是所使用的调查问卷的异质性,因此,选择恰当有效的问卷是非常重要的。生活质量的测量应该是多维的,至少包括三个可能受疾病或治疗影响的广泛领域,包括身体、心理和社会功能[26]。Hartman等[27]在对320名肛肠畸形和HD患者的研究中发现,主要影响成人巨结肠患者生活质量的不是身体症状,而是心理和社会功能。生活质量对成年人的社会、心理影响虽然已经引起人们的广泛关注[28-30],但至今尚未形成系统、统一评价标准。Svetanoff等[31]研究发现,在ARM和HSCR患者的生活中存在多种社会、心理压力,为ARM和HSCR患者及其家庭提供与成长相匹配的医疗、心理和社区支持,可改善生活质量。对HSCR患者进行术后随访并提供相应的帮助非常重要,该例患者在术后1个月电话随访进行生活质量评估时发现,患者对手术及身体康复方面较为满意,但在社会活动、心理状态方面存在一些问题,需要建立长期随访机制对患者进行疏导,帮助患者逐渐改善生活质量。
综上所述,ARM合并HSCR是一种先天性消化道畸形,临床上非常少见,成年患者罕见。而ARM患者往往合并其他器官的畸形,当在临床诊疗中发现ARM时,一定要引起高度重视,务必考虑合并其他器官畸形可能,必要时进行详细检查,对HSCR尤需警惕。此外,对HSCR患者的手术选择不应拘于传统常规手术,应根据不同患者疾病特点,制定个体化治疗方案,灵活选择对患者有益而且医师擅长的术式。最后,手术的目的是去除疾病、改善患者生活质量,对于术后患者尤其是永久性结肠造口患者,其存在一定的心理负担,医务人员应关注患者术后生活质量,予以心理支持,有助于患者获得良好的远期生活质量。
