Adsorption and rotational barrier for a single azobenzene molecule on Au(111)surface∗
2021-09-28DongHao郝东XiangqianTang唐向前WenyuWang王文宇YangAn安旸YueyiWang王悦毅XinyanShan单欣岩andXinghuaLu陆兴华
Dong Hao(郝东),Xiangqian Tang(唐向前),Wenyu Wang(王文宇),Yang An(安旸),Yueyi Wang(王悦毅),Xinyan Shan(单欣岩),and Xinghua Lu(陆兴华),3,4,‡
1Beijing National Laboratory for Condensed-Matter Physics,Institute of Physics,Chinese Academy of Sciences,Beijing 100190,China
2School of Physical Sciences,University of Chinese Academy of Sciences,Beijing 100190,China
3Songshan Lake Laboratory for Materials Science,Dongguan 523808,China
4Center for Excellence in Topological Quantum Computation,Beijing 100190,China
Keywords:Au(111),molecular rotor,azobenzene
Molecular motors and rotors are potential building blocks for ultimate small mechanical devices in the future.[1–7]Fundamental physical properties of such molecules have been the focus in many studies nowadays.For example,the dynamic behaviors of the molecules were investigated when they were adsorbed onto the surface and visualized by scanning tunneling microscopy(STM).[8–11]Au(111)is a common surface in these studies due to its inertial chemical characteristics and easy sample preparation.One of its unique features is the complex herringbone surface reconstruction.[12–19]The alternative fcc and hcp regions in the herringbone reconstruction create periodic electronic quantum wells on the surface by which the electronic surface state and adsorption potential are modulated.[20]The kink sites of the herringbone reconstruction are attractive points for many molecules,such as benzene,[21]water,[22]carbon monoxide,[23]etc.Besides,they act as perfect anchor points or pivots for the molecular rotors in various control experiments where the rotation of the molecules can be excited by tunneling current,[24]voltage pulse,[8]and/or laser illumination.
Our recent experiment on dynamic behavior of individual azobenzene molecules on Au(111)surface revealed distinct roles in rotation excitation by tunneling electrons and ultrafast laser pulses.[25]As shown in Fig.1,the azobenzene molecules adsorbed on to the kink sites present two equivalent ground states separated by 60°.Orientation switching between these two states occurs when the molecules are excited by electrons and/or photons.While it has been revealed in the paper that there is an energy barrier between the two orientations,the magnitude of such barrier was not well quantified yet.The detailed adsorption configuration has not been revealed either,due to the lack of atom-resolved topographic images.To make a step forward,here we report the investigation on the adsorption configuration and rotational dynamics for this molecular rotor by first-principles density functional theory(DFT)computation.The anchor site is revealed by comparison among various possible adsorption configurations of a benzene ring on the surface,and the rotational energy barrier is obtained by sweeping the rotational angle of an azobenzene molecule with one of its phenyl rings fixed at the kink site.The validity of the computational results is supported by the comparison with the experimental data.
The periodic DFT calculations were performed using the pseudopotential method and plane-waves as implemented in the Quantum ESPRESSO code,version 6.4.1.[26]The generalized gradient approximation(GGA),Perdew–Burke–Ernzerhof(PBE)exchange–correlation functional,[27]and projector-augmented wave(PAW)pseudopotentials[28]were employed throughout.Cutoffs for the expansion of the plane waves and charge density were chosen according to the SSSP pseudopotential verification database,[29]i.e.,650 eV and 5000 eV,respectively.Spin polarization was not considered in these adsorption systems.The Brillouin zones were sampled by using the Monkhorst–Pack methodology,[30]withΓ point-centered 3×3×1 grids for relaxations and 5×5×1 grids for the total energy calculations.Partial occupancies of electronic states were set according to the method and system.For relaxations involving fcc gold lattice only,the Methfessel–Paxton method[31]with a smearing width of 0.30 eV was used.A Gaussian smearing with a width of 0.05 eV was adopted throughout the relaxations of all the composed systems.To calculate the total energies,the Gaussian smearing method with the width of an order of magnitude smaller was employed.The self-consistent field(SCF)calculation convergence threshold was set to 7×10−6eV for all calculations.For the geometry optimizations,a 6.5×10−3eV/˚A force convergence criterion was applied;during the single point calculations the forces were checked to ensure that the threshold was not exceeded.
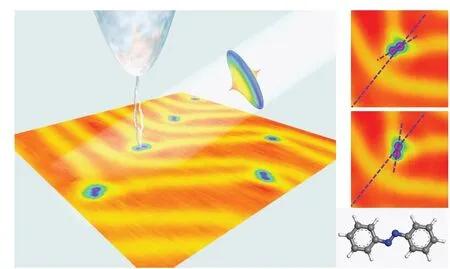
Fig.1.Azobenzene molecules on Au(111)surface.The molecules are adsorbed onto the kink site of the herringbone surface reconstructions and present two orientation states,±30° with respect to the line bisect the herringbone reconstruction.The molecules can be excited to toggle between the two orientations,by tunneling electrons and/or ultrafast laser pulses.
The molecule–surface system is modeled in supercell geometries employing the GGA-PBE-optimized lattice constants.Depending on the registry of the aromatic molecule with the substrate,unit cells with sizes of(4×4×4)and(6×5×4)are established for the adsorption of benzene and azobenzene,respectively.The system is padded by 20˚A of vacuum in the z-direction,and a dipole correction is applied to offset the effect of an artificial electric field due to the asymmetric slab structure.The supporting gold substrate is four atomic layers thick with the two bottom layers kept fixed for calculating benzene on the surface.The semi-empirical Grimme-D3 dispersion correction[32]is applied to describe the long-range van der Waals(vdW)interaction in the systems.This version of DFT+D correction scheme was compared with other empirical dispersion corrections by Werner Reckien et al.[33]and with the vdW-DF method by Karen Johnston et al.,[34]which exhibits a good agreement with the experimental data[35]for benzene adsorption on coinage metals.
We first relax the Au(111)crystal and an isolated benzene molecule separately to obtain the Au lattice constant and atomic structure of the benzene molecule.To determine the optimal structure and calculate the energy of an isolated molecule using periodic DFT,the molecule is placed in a large enough cubic supercell with the side length of 15˚A to minimize the influence from its periodic images.To perform the geometric optimization,all internal degrees of freedom like bond lengths and bond angles are taken into considerations with appropriate stopping criteria for the energy and force.We start from various initial structures with all degrees of freedom included to check whether they converge to the same structure to ensure that the optimal structures are obtained.Figure 2 shows the structural and energy convergence of both the Au crystal and the isolated benzene molecule.The Au–Au distance is stabilized to 2.941˚A,and the C–C bond length of benzene changes converges to 1.397˚A.The calculated final structural parameters are employed in our subsequent calculations.
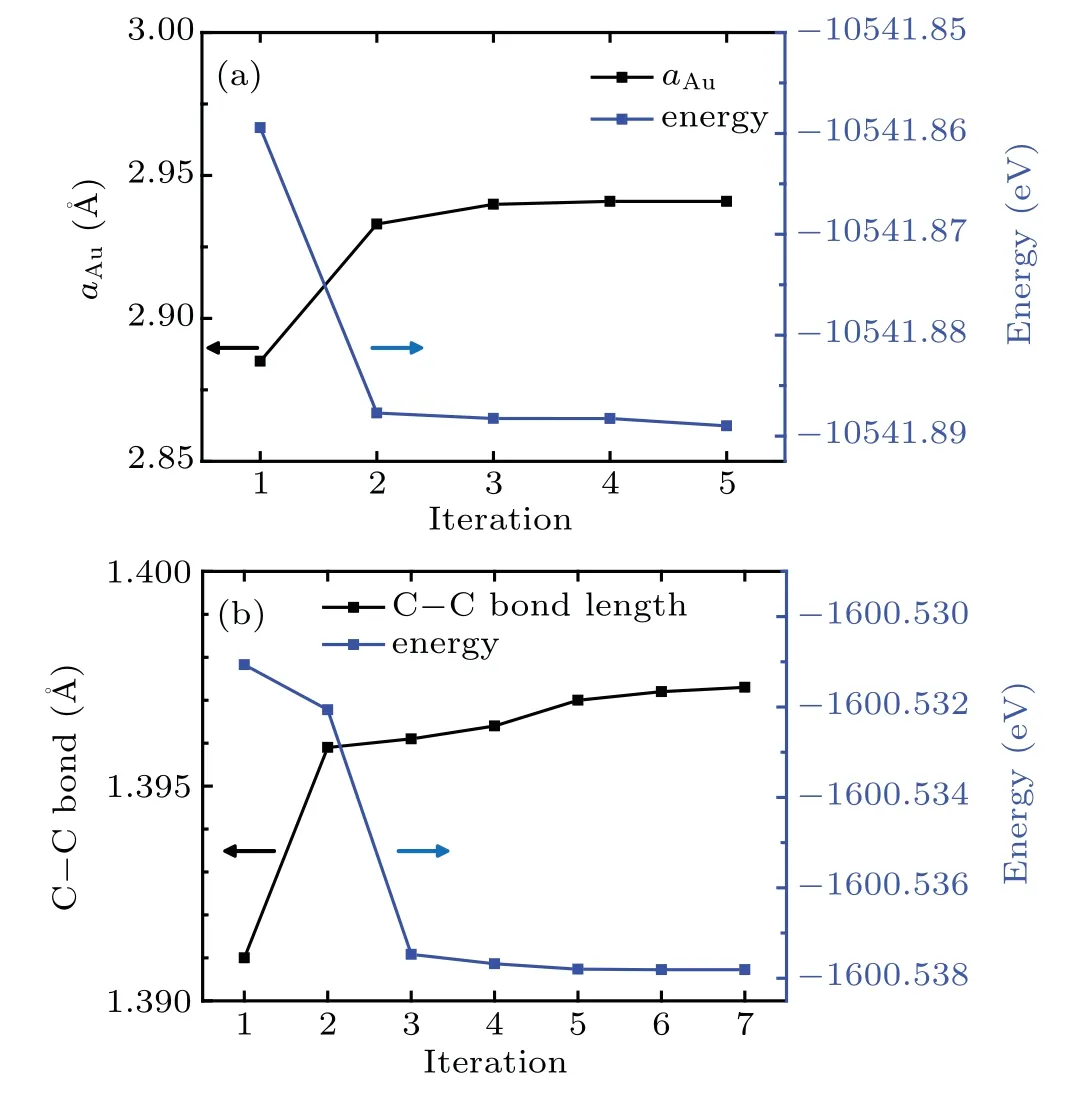
Fig.2.QE calculation set up.Both the Au(111)surface and the isolated benzene molecules are relaxed to their ground energy states.A unit cell consisting of 4×4×4 Au atoms is used and the Au–Au distance at equilibrium is 2.941˚A.The C–C distance in benzene molecule is calculated to be 1.397˚A in its ground state.
The ground state for a molecule on surface is obtained by comparison among all possible configurations of the adsorption system.The adsorption energy Eadsis defined as
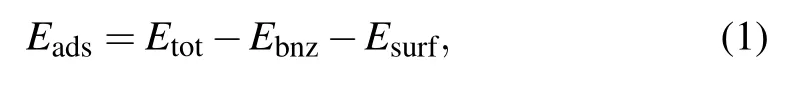
where Etot,Ebnz,and Esurfare the energies of the whole system,the isolated optimized molecule,and the bare metal surface,respectively.Figures 3(a)and 3(b)show the possible adsorption sites on the close packed Au(111)surface.The top site(T)is above a surface gold atom,the bridge site(B)is midway between adjacent top sites,the fcc and hcp hollow sites(Ha and Hb)are situated in the midpoint of three top sites,as shown in Fig.3(a).Geometry optimizations are carried out on each high-symmetry site to obtain the distance between the center of the benzene molecule and the atom in the top surface layer,as shown in Fig.3(b).As another degree of freedom for the adsorbed molecule,the orientation is defined as the angle between one of the C–H bonds and the crystal a-axis,as shown in Fig.3(a).Considering the six-fold symmetry of the surface,two typical orientations,0°and 30°,are examined in our study.

Fig.3.(a)The adsorption configuration of the benzene molecule on fully optimized Au(111)surface.The top layer Au atoms are the goldcolored spheres,the second layer is beige,and the third layer is represented by the nut-brown spheres.The vectors a and b in the bottom left are the crystallographic axes.Eight configurations are examined:top site(T),bridge site(B),fcc and hcp hollow sites(Ha and Hb),respectively,with two orientations for each,i.e.,0°and 30°(see the unit cell in the bottom right).(b)Schematic cross-sectional view for a benzene molecule above an fcc hollow site of the Au(111)surface.For all configurations,the molecule–surface distance is calculated from the center of the benzene molecule to the gold atom in the top layer.(c)Calculated adsorption energy as well as height Z for all configurations in this study.The inset shows the most stable adsorption registry on the fcc hollow site.(d)The relative energy variation for orientations near 30° for the registry of a benzene molecule on the fcc hollow site.The minimum at 30° confirms the preferred adsorption configuration.
The lowest energy states for each configuration are shown in Fig.3(c).Among all geometries,the values of the adsorption energy and height for the top site are apparently larger than those for the hollow and bridge sites.The maximum energy difference between different sites is about 85 meV.For adsorption on the hollow and bridge sites,the benzene molecule is oriented so that one of the C–H bonds makes an angleα=30°with the a-axis.In contrast,the molecule on the top site prefers an orientation with the angleα=0°;that is,the C–H bond of benzene is parallel to the Au(111)a-or b-axis.The adsorption height is~3.12˚A for both the bridge and hollow sites,whereas for the top site it is 0.1˚A larger(~3.22˚A).The value of 3.12˚A is in good agreement with the results of Werner Reckien et al.[33]and Javier Carrasco et al.[36]The preferred adsorption configuration for a benzene molecule is the fcc hollow site with the orientation angle of 30°.
The nonbonded vdW interaction between the adsorbate and the surface is of crucial importance in such calculations.Without vdW correction,the molecular height Z is 0.3˚A higher than that on the fcc hollow site(Ha-30°).It is clear that the interaction between benzene and gold surface can be mostly attributed to the vdW forces.This is not surprising since both the benzene molecule and the gold substrate have relatively high polarizabilities as already known by experiment[21]and theoretical studies.[34]Figure 3(d)shows the variation of adsorption energy under the perturbation of molecular orientation near 30°.The minimum at the angle of 30°confirms that it is the most stable configuration,in which three carbon atoms in the phenyl ring locate nearly on top of gold atoms.
Next we examine the adsorption behavior of azobenzene molecule on the Au(111)surface.To simulate the anchor on Au(111)surface,we place the azobenzene molecule on the surface with the center of one of the phenyl rings above the fcc hollow site,the stable adsorption site for benzene molecules.The adsorption heights of the molecule at a specific orientation are estimated by calculating the minimum adsorption energy as a function of molecule–substrate distance.The orientational angle is defined by the angle between the line connecting centers of two phenyl rings and the crystallographic a-axis,as shown in the inset of Fig.4.Five orientational angles from 10°to 50°are investigated,and all derive minimum adsorption energy at the adsorption height of 3.25˚A,as shown in Fig.4.This value gives a reasonable accuracy as compared with the experimental value of 2.97±0.05˚A and the calculated value of 3.5˚A using the revPBE-vdW-DF functional.[37]
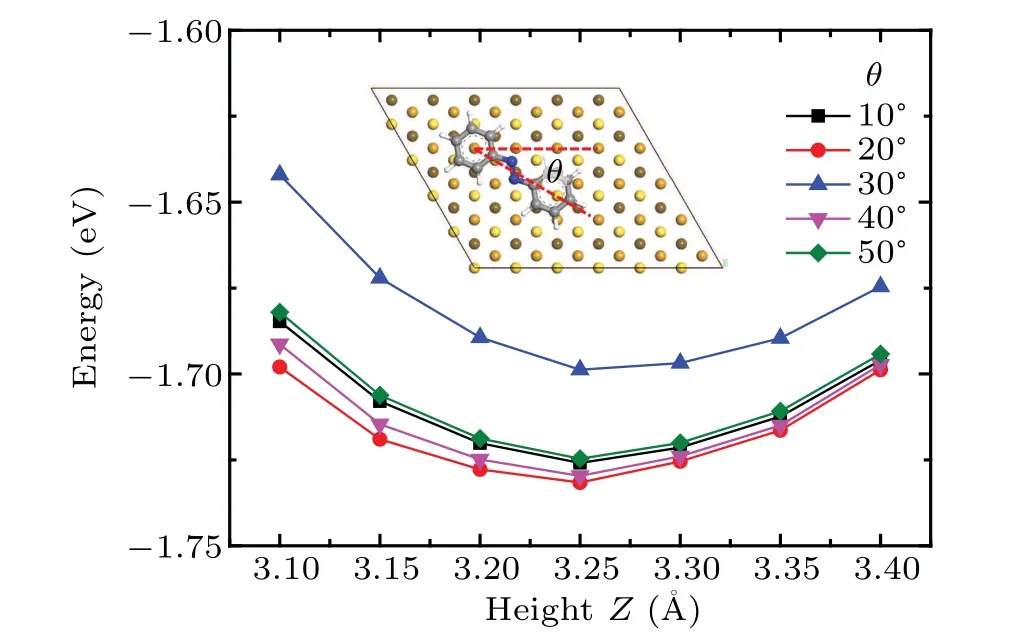
Fig.4.The adsorption energy of azobenzene molecule on Au(111)surface as a function of the molecule–surface distance,under various adsorption orientations.Inset:the adsorption configuration of the azobenzene molecule on the fully optimized Au(111)substrate with definition of orientation angleθ.The color code for atoms is the same as used in Fig.3.
Finally,the rotational barrier of an azobenzene molecule anchored at the kink site is obtained by examining the rotational energy potential as a function of molecular orientation.With the adsorption height fixed at 3.25˚A,the rotational energy potential curves are obtained by single-point calculations with the PBE-D3 approach.We stepwise alter the molecular orientationθfrom 0°to 60°,and the obtained energy profiles are shown in Fig.5.The computed adsorption energy is thus about 1.76 eV,and the rotational energy barrier is about 50 meV.The results are on the right order of magnitude with the experimental data(24 meV)[25]and those of azobenzene derivatives(33±3.8 meV).[40]We also tried single-point calculations using the vdW-DF method,the known nonlocal vdW density functional,and acquired similar results.In Fig.5(b),we plot the vdW surface maps of the molecular system at two different orientations.The vdW surface maps are usually set by the electron density of the isosurfaces as of 0.002±δelectrons/Bohr3,whereδis the tolerance.Such isosurfaces enclose approximately 95% of the molecular electron density.We also plotted the isosurfaces with the electron density set as 0.010 electrons/Bohr3.For both orientations,the isosurfaces of carbon atoms in the anchored phenyl ring have remarkable overlap with those of underlying Au atoms.The sliding phenyl ring shows more overlap at orientationθ=0°than forθ=30°,indicating a weaker vdW energy atθ=30°.This is consistent with the rotational energy barrier profile.The isosurface of the nitrogen atoms,on the other hand,shows much less overlap with the substrate,suggesting a much weaker interaction with the substrate.
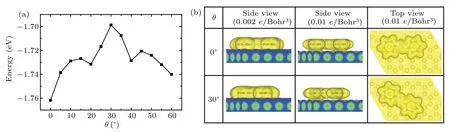
Fig.5.(a)The adsorption energy as a function of orientation for an azobenzene molecule with one of its phenyl rings pivots around the fcc hollow site.(b)van der Waals surface map of the molecular system with the electron density of the isosurface being 0.002 electrons/Bohr3(left column)and 0.01 electrons/Bohr3(middle and right columns).
We note that the computational results are not in perfect match with the experimental results.The computed adsorption energy of 1.76 eV is about 20%lower than the estimated value from experiment(~2.1 eV).The difference arises from the inadequate model for the kink site of herringbone reconstruction.Calculation on the herringbone surface reconstruction including the kink site,unfortunately,is extremely difficult since the whole herringbone pattern consists of more than 1000 atoms in a single layer.In addition,employing pairwise dispersion-correction scheme,such as the PBE-D3 scheme,usually leads to a certain degree of overestimation in interaction energies for molecules on surfaces.[38]The overbinding is sizeable even with effectively included substrate screening via vdWsurf(PBE+vdWsurf).[39]The sources of error are not only the intrinsic limitation of the Grimme dispersion correction—insufficient response to the local chemical environment and collective response effects along with the absence of higher-order dispersion terms,but also the entangled finitetemperature effects due to the pronounced element of flexibility,disorder,and structural anharmonicity in the targeted molecules.Nevertheless,the adsorption energy profile for the rotation of azobenzene on the Au(111)surface has depicted the shapes of the potential well in general,and the computed energy barrier of 50 meV is on the same order as that estimated from experimental data.Further improvement is possible with advanced computation facility as well as new development in theoretical methods.
In summary,we investigated the adsorption configurations and rotational behavior for single azobenzene molecules on Au(111)surface.The adsorption configuration for a benzene molecule on the surface was calculated to illustrate the anchoring effect on the kink site of herringbone surface reconstructions.The potential energy profile has been calculated to reveal the rotation barrier for the molecule that is in agreement with the experimental data.The approach as demonstrated in this study is applicable to a broad spectrum of molecular systems.
猜你喜欢
杂志排行

Chinese Physics B的其它文章
- Origin of anomalous enhancement of the absorption coefficient in a PN junction∗
- Protection of isolated and active regions in AlGaN/GaN HEMTs using selective laser annealing∗
- First-principles study of plasmons in doped graphene nanostructures∗
- Probing thermal properties of vanadium dioxide thin films by time-domain thermoreflectance without metal film∗
- An improved model of damage depth of shock-melted metal in microspall under triangular wave loading∗
- Signal-to-noise ratio of Raman signal measured by multichannel detectors∗