猪流行性腹泻病毒S蛋白纳米抗体的筛选与鉴定
2021-09-27王天宇李志伟董林芳马志倩边巴次仁肖书奇
王天宇,李志伟,杨 婷,董林芳,马志倩,边巴次仁,肖书奇,李 爽*
(1.西北农林科技大学动物医学院,杨凌 712100;2.西藏阿里地区中等职业技术学校,阿里 859000)
猪流行性腹泻(porcine epidemic diarrhea,PED)是一种接触传染性肠道病毒病,以仔猪的水样腹泻、高死亡率和发病率为特征[1-2]。PED的病原猪流行性腹泻病毒(porcine epidemic diarrhea virus,PEDV)是一种单股正链RNA病毒,基因组大小约28 kb,编码16个非结构蛋白(non-structural protein, nsp),4个结构蛋白(S、E、M和N)和1个辅助蛋白ORF3[3]。在结构蛋白中,S蛋白根据功能划分为S1和S2,S1结构域有助于受体识别、引发肠致病性和免疫原性,因为它包含显性中和表位。S2结构域锚定在病毒膜上,触发膜融合[4]。S1蛋白中的中和表位分别为COE(499—638 aa)、SS2(748—755 aa)和SS6(764—771 aa),(1 368—1 374 aa)位于S2结构域中[5-7]。除此之外,S1蛋白的N端(34—230 aa)含有比较重要的抗原表位,可以诱导保护性反应,负责与细胞表面唾液酸受体结合[8]。此外,S蛋白是PEDV的一个主要的毒力基因[9-10],也是分析PEDV遗传变异和分子流行病学的重要靶标[11],这使得S蛋白成为研发疫苗和诊断试剂的候选者。
20世纪90年代,一群比利时学生尝试纯化单峰骆驼血清中的蛋白,无意中发现了除传统抗体之外,还有一种缺少轻链,仅由重链组成的抗体,这种抗体后来被称为重链抗体[12]。随后,在羊驼和鲨鱼等动物外周血中也发现了这种特殊抗体的存在。其重链可变区组成的单域抗体称为VHH(variable domains of Camellidae heavy chain-only antibodies,VHH)。VHH的形状为扁长型,类似于橄榄球状,相对分子质量约为15 ku,直径约为2.5 nm,高约4 nm,VHH是已知最小的具有完整抗原结合位点的抗体,因此被称为纳米抗体(nanobody,Nb)[13]。与常规抗体相比,Nb稳定性高,可溶性好、亲和力强、免疫原性小及易在大肠杆菌中表达和纯化,且具有高表达产量等特点[14-15]。因此,这使得Nb被应用到许多医学、生物技术、诊断和治疗中[16-17]。
目前,还没有报道针对PEDV S蛋白纳米抗体用于PED的治疗和诊断中,本研究拟通过噬菌体展示技术筛选出PEDV S1蛋白的特异性纳米抗体,为PED的治疗、诊断及研究PEDV的致病机制奠定基础。
1 材料与方法
1.1 材料
E.coliTrans5α和E.coliBL21(DE3)感受态细胞均购自上海唯地生物技术有限公司;pCANTAB 5E噬菌体载体购自美国GE公司;HRP-Goat@Mouse IgG抗体和Anti-His Mouse mAb均购自于Jackson immuoresearch公司。Ficoll-Paque PLUS淋巴细胞分离液购自Greiner bio-one公司;辅助噬菌体M13K07和限制性内切酶购自美国NEB公司;RNAiso Plus、PrimeScript RT reagent Kit和Prime STAR HS DNA polymerase购自日本TaKaRa公司;质粒提取试剂盒和胶回收试剂盒购自北京全式金生物科技有限公司。
1.2 方法
1.2.1 重组S1蛋白原核表达载体的构建 用RNAiso Plus从PEDV(GenBank序列号: No MT787025)阳性的组织病料中提取RNA,用反转录试剂盒将RNA反转成cDNA, 用表1引物以cDNA为模板扩增S1基因(66—1 515 bp),通过BamH Ⅰ和Hind Ⅲ 酶切位点克隆到pET32a载体中,得到质粒pET32a-S1,测序正确后,用于后续试验。使用EasyPure Plasmid MiniPrep Kit进行 pET32a-S1的质粒提取,检测质粒浓度,置于-20 ℃保存。

表1 构建重组S1蛋白原核表达载体的引物序列
1.2.2 重组S1蛋白的表达与纯化 将pET32a-S1质粒阳转到E.coliBL21(DE3)感受态细胞中,挑取阳性克隆到LB培养基中,于37 ℃恒温摇床中220 r·min-1震荡培养过夜,按照1∶100的比例接种于大的摇瓶中,培养至对数期,加入IPTG贮存液(1 mol·L-1)至终浓度为1 mmol·L-1,诱导5 h后收菌,SDS-PAGE鉴定蛋白是否表达。对诱导后的菌体138 kW超声3 s,暂停3 s,共超声40 min,收集上清和沉淀进行蛋白可溶性分析。用Ni-NTA纯化S1蛋白。
1.2.3 动物免疫 5 mg纯化的PEDV S1蛋白同弗氏完全佐剂和不完全佐剂乳化后免疫阿拉善双峰驼。免疫间隔为2周,4次后,采用间接ELISA测定骆驼血清中特异性PEDV S1抗体滴度,随后采集骆驼外周抗凝血,并分离淋巴细胞。间接ELISA的具体操作步骤:使用PBS作为包被缓冲液,将截短PEDV S蛋白稀释至10 μg·mL-1,100 μL·孔-1,加入酶标板,4 ℃条件包被过夜;次日,用PBS’T洗板 4次,200 μL·孔-1加入2.5%脱脂奶粉,37 ℃恒温孵育1 h;弃去封闭液,用PBS’T洗板4次,对骆驼血清样品使用封闭液进行倍比稀释,各稀释比例100 μL·孔-1,置于37 ℃恒温孵育1 h;弃去倍比稀释的骆驼血清样品,PBS’T洗板4次,使用封闭液1∶2 000 稀释Rabbit @ Camel IgG多克隆抗体,100 μL·孔-1,37 ℃恒温孵育1 h;弃去抗血清,PBS’T洗板4次,100 μL·孔-1加入稀释至工作浓度的Goat @ Mouse IgG HRP标记抗体,37 ℃恒温孵育1 h。弃去酶标二抗,用PBS’T洗板4次,100 μL·孔-1加入TMB显色底物,37 ℃避光恒温孵育15 min;50 μL·孔-1加入3 mol·L-1H2SO4,使用分光光度计测量OD450 nm。
1.2.4 噬菌体展示文库的构建 提取外周血淋巴细胞RNA并反转录成cDNA.采用巢式PCR扩增VHH基因,用表2中CALL-1和CALL-2引物扩增出大小约700 bp的片段; 随后,用表2中VHH-F和VHH-R进行第二轮的扩增,大小约400 bp, 对第二轮的产物回收,对回收产物和pCANTAB 5E载体同时进行双酶切,酶切位点为PstⅠ和NotⅠ;随后将载体与目的片段连接并电转入E.coliTG1感受态细胞中,将转化的感受态细胞在37 ℃ 120 r·min-1恒温培养40~60 min。吸取200 μL培养物用于所构建文库质量的鉴定,将剩余培养物1.5 mL·板-1均匀涂布至 LB/AMP-GLU方形培养皿,37 ℃恒温箱中培养6~8 h。刮下培养皿内菌落到2~3 mL LB/AMP-GLU培养基,保存备用。

表2 VHH片段PCR扩增引物
1.2.5 文库容量和多样性测定 将上述步骤中取出的200 μL培养物10倍梯度稀释,并涂布到AMP-GLU LB平板,37 ℃恒温培养过夜。统计转化子数量。挑取96个单菌落并菌液PCR鉴定阳性率,对阳性克隆测序;使用MegAlign软件对VHH序列进行比对,分析文库的序列多样性。
1.2.6 VHH噬菌体抗体展示文库的救援 取“1.2.4”步骤中构建的VHH噬菌体抗体展示文库,加入100 mL 2×YT/AMP-GLU培养基,37 ℃剧烈震荡培养至对数生长期。加入20 MOI辅助噬菌体M13KO7,37 ℃ 200 r·min-1震荡培养1~2 h,收集菌体沉淀。沉淀重悬后继续震荡培养12 h。离心,收集上清,加入1/5体积预冷的PEG/NaCl溶液,冰中孵育10~12 h。离心后用PBS重悬后即为重组噬菌体。
1.2.7 PEDV S1特异性重组噬菌体的淘选 用PEDV S1蛋白包板,100 ng·孔-1,同时设置对照孔,4 ℃包被过夜。弃包被液,2.5%脱脂奶粉封闭,37 ℃孵育1 h;1012pfu·mL-1加入步骤“1.2.6”救援的重组噬菌体,37 ℃孵育1 h。弃噬菌体溶液,洗板后加入0.1 mol·L-1的三乙胺,室温静置10 min,收集洗脱液,立即加入相同体积的1 mol·L-1Tris-HCl(pH7.4),随后对所洗脱的重组噬菌体进行滴度的测定。取20 mL培养至对数期的TG1,加入洗脱的噬菌体溶液,37 ℃静置侵染60 min后加入80 mL 2×YT/AMP-GLU培养基,37 ℃ 200 r·min-1,震荡培养至对数期,按照上述步骤加入20 MOI辅助噬菌体进行救援后进行二轮淘选;按照上述步骤,对重组噬菌体总共进行3轮固相淘选。
1.2.8 PEDV S1特异性重组噬菌体富集 以PEDV S1包板过夜。封闭后将重组噬菌体溶液使用封闭液按1∶10稀释,37 ℃孵育1 h;弃去噬菌体溶液,PBS’T洗板 4次,加入 HRP标记Mouse @ M13噬菌体抗体,37 ℃孵育1 h。弃去抗体,PBS’T洗涤4次,加入TMB显色底物,37 ℃孵育15 min。50 μL·孔-1加入3 mol·L-1H2SO4终止显色,测量OD450 nm。
1.2.9 重组纳米抗体的表达 取第3轮淘选后的重组噬菌体溶液,重组噬菌体溶液十倍梯度稀释后,加到对数期的TG1,37 ℃感染60 min,涂布于LB/AMP-GLU,过夜培养;挑取单菌落到LB/AMP-GLU中,培养过夜;从各菌落培养物中取出50 μL分别转接于2 mL TB培养基中,对应编号置于24孔培养板,培养至对数期;加入终浓度为1 mmol·L-1IPTG,过夜诱导表达。
1.2.10 重组纳米抗体粗提物的制备 收集步骤“1.2.9”中的24孔板中培养液于1.5 mL EP管中,离心收集菌体沉淀,加入PBS重悬菌体并-20 ℃冻融。离心后,上清即为纳米抗体粗提物。
1.2.11 重组纳米抗体粗提物的检测 将经过纯化的截短PEDV S1重组蛋白用PBS缓冲液稀释至100 μg·mL-1,100 μL·孔-1,4 ℃包被过夜,同时设置相同数量的对照孔,包被等量的PEDV N重组蛋白;进行封闭后,用PBS’T洗涤4次,加入使用封闭液按1∶1稀释的纳米抗体粗提物,100 μL·孔-1,37 ℃ 恒温孵育1 h;弃去粗提物溶液,用PBS’T洗板4次,加入使用封闭液按1∶2 000稀释至工作浓度的Anti-E-tag标签抗体,100 μL·孔-1,37 ℃恒温孵育1 h;弃掉Anti-E-tag标签抗体,用PBS’T洗涤4次,100 μL·孔-1加入使用封闭液稀释至工作浓度的HRP标记Goat@Rabbit IgG抗体,37 ℃ 恒温孵育1 h;随后进行显色,进行使用分光光度计测量OD450 nm。
1.2.12 PEDV S1特异性纳米抗体的序列分析 将上述ELISA鉴定为阳性的克隆测序。用MegAlign软件比对测序结果,并根据纳米抗体高变区分类。
1.2.13 PEDV S1蛋白纳米抗体的特异性和结合力测定 使用同期对骆驼免疫的2种蛋白,猪圆环病毒Cap蛋白和猪伪狂犬病病毒gE蛋白作为对照。将截短PEDV S蛋白、猪圆环病毒Cap蛋白和猪伪狂犬病病毒gE蛋白包被于同一个ELISA酶标板,利用“1.2.11”步骤检测所筛选PEDV S蛋白纳米抗体的特异性。对不同的纳米抗体粗提物10倍倍比稀释,用同样的方法测定纳米抗体的结合力。
1.2.14 纳米抗体活性的鉴定 将生长状态良好的Vero 细胞以一定密度铺入含有24孔爬片细胞板中和正常的12孔细胞培养板中,24 h后接入0.1 MO1 PEDV;接毒后24 h,一方面进行间接免疫荧光检测;每孔加入200 μL 4%多聚甲醛,置于37 ℃恒温箱中固定15 min;弃掉固定液后,每孔加入200 μL 0.25% triton X-100,15 min后取出。用1×PBS(K+)洗涤3次,加入1%BSA封闭30 min;加入原核表达的纳米抗体Nb3,37 ℃孵育1 h;洗涤后加入His抗体,37 ℃孵育1 h;洗涤后加入荧光二抗,孵育1 h;洗涤后加入DAPI,染色10 min,使用细胞成像Leica microscope进行拍照。另一方面进行Western blot: 收集12细胞培养板中正常Vero细胞和PEDV感染的Vero细胞。利用NP40裂解细胞,取10 μL进行SDS-PAGE;随后转印至PVDF膜,用5%脱脂奶粉封闭2 h,用Nb3室温孵育1 h,PBS’T洗涤4次后,加入His 抗体室温孵育1 h,PBS’T洗涤4次后,用HRP标记的Goat anti-Mouse IgG(H+L)二抗进行室温孵育1 h,PBS’T洗涤4次后,将PVDF膜置于ECL发光液中后,放入化学发光成像仪内进行成像。
2 结 果
2.1 pET32a-PEDV-S1原核表达载体的构建
提取PEDV阳性组织的RNA,反转录成cDNA,以此为模板,PCR扩增产物位于1 000~2 000 bp,与预期目的片段大小(1 452 bp)相符(图1A),目的条带回收后与载体pET-32a同时双酶切(图1B),连接转化,挑取单菌落进行PCR鉴定,结果如图1C, 将阳性克隆送公司测序,综上结果表明,成功构建pET32a-PEDV-S1原核表达载体。

A.PEDV S1基因的扩增;B.目的片段和载体的双酶切;C.菌液PCR鉴定
2.2 PEDV S1蛋白的表达与纯化
将测序正确的pET32a-PEDV-S1质粒转到E.coliBL21(DE3)中,以1 mmol·L-1的IPTG诱导表达后SDS-PAGE检测,在大约70 ku处可见与预期结果一致的条带,而空载诱导后,未出现此条带(图2A),且S1 重组蛋白以包涵体的形式存在(图2B)。用Ni-NTA纯化后,得到纯度较好S1重组蛋白(图2C)。
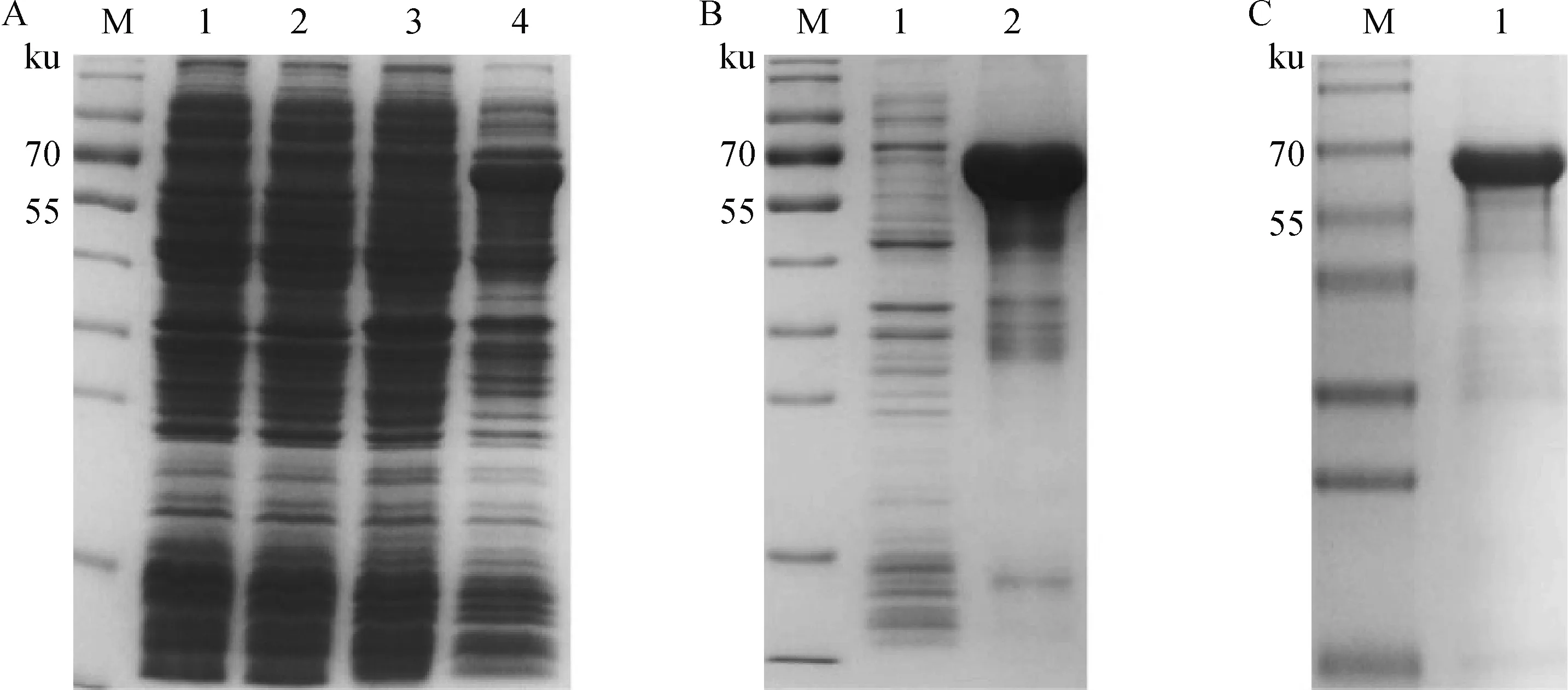
A.PEDV S1重组蛋白的诱导表达(M.蛋白质相对分子质量标准;1.pET-32a空载体未诱导菌体;2.pET-32a空载体诱导菌体;3.pET-32a-S1表达载体未诱导菌体;4.pET-32a-S1表达载体诱导菌体);B.PEDV S1重组蛋白的可溶性鉴定(M.蛋白质相对分子质量标准;1.菌体裂解后上清;2.菌体裂解后沉淀);C.PEDV S1重组蛋白的纯化
2.3 双峰驼免疫
用纯化后的截短PEDV S重组蛋白免疫阿拉善双驼峰,在最后一次免疫后第4天,用间接ELISA检测S蛋白抗体效价,结果显示,双驼峰血清中PEDV S抗体效价高达1∶256 000(图3)。

图3 ELISA检测骆驼血清中PEDV S1特异性抗体效价
2.4 VHH噬菌体展示文库的构建
采集200 mL骆驼外周抗凝血,分离外周血淋巴细胞。提取RNA,反转录成cDNA。通过巢式PCR扩增VHH片段,第一轮回收700 bp左右目的条带(图4A),第二轮PCR回收 400 bp左右条带(图4B)。将VHH片段构建至pCANTAB 5E载体中,电转化至TG1感受态得到噬菌体展示文库,该文库库容约为2.1×107,阳性率约为85%(图4C)。

A和B.扩增VHH基因;C.VHH文库的阳性率鉴定
2.5 PEDV S1蛋白特异性纳米抗体的筛选
对噬菌体展示文库进行救援后,经过3轮淘选后,富集率可以达到1 673(图5A)。将第3轮淘选后的重组噬菌体侵染对数期 TGI细胞,涂布至 LB/AMP-GLU平板,挑取96个单菌落,共挑取2轮,进行粗提,通过ELISA鉴定粗提物与PEDV S蛋白的反应性,结果如5B,随机选取的96个单克隆中有23个为阳性克隆,对阳性克隆进行测序分析,鉴定出了6个序列不同的纳米抗体,并依次命名为Nb1~Nb6(图5C)。

A.特异性噬菌体富集结果;B.间接ELISA筛选PEDV S1蛋白特异性纳米抗体;C.PEDV S1蛋白特异性纳米抗体的氨基酸序列比对
2.6 PEDV S蛋白纳米抗体特异性及结合力的测定
以猪圆环病毒Cap重组蛋白和猪伪狂犬病病毒gE重组蛋白作为对照,检测筛选出的6株纳米抗体的特异性。结果显示,6株纳米抗体均不存在交叉反应(图6A)。与此同时,通过ELISA进一步确定了6株纳米抗体对PEDV S重组蛋白的结合能力,结果显示,Nb3具有最高的结合能力(图6B)
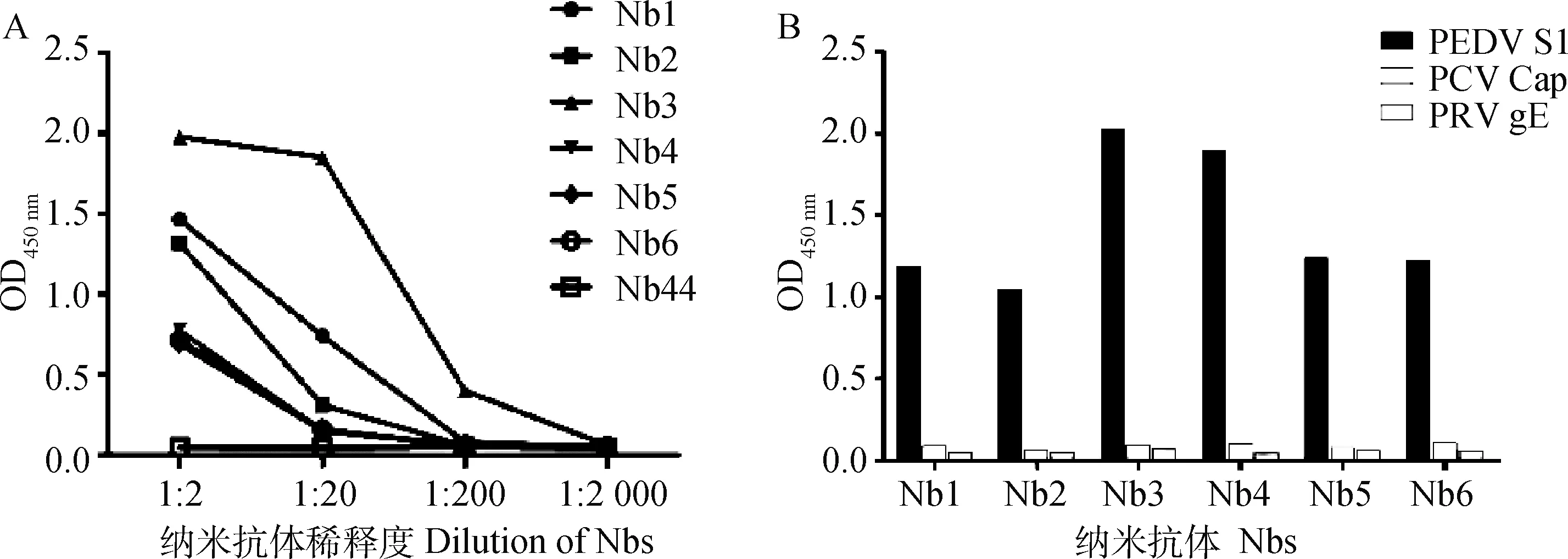
图6 PEDV S蛋白纳米抗体的结合力(A)与特异性(B)的分析
2.7 Nb3与PEDV结合活性鉴定
通过Western blot和IFA验证纳米抗体Nb3与PEDV的结合,用原核表达的纳米抗体Nb3验证其是否能够结合感染细胞中的PEDV,以未接毒细胞作为对照,结果如图7,Nb3同 PEDV具有良好的结合活性。

A.WB鉴定纳米抗体Nb3与PEDV的结合(1.PEDV 感染Vero细胞;2.正常Vero细胞):B.间接免疫荧光鉴定纳米抗体Nb3与PEDV的结合(标尺=200 μm)
3 讨 论
2010年10月,PED在中国大面积暴发,这次疫病的暴发以7日龄以内的哺乳仔猪极高发病率和死亡率为主要特征,2周龄以上的猪出现不同程度的腹泻和厌食等症状,并预示着高毒力PEDV变异毒株的出现[19],由此可见,PEDV是一种破坏力较大的肠道冠状病毒[19-21],目前,尚无针对 PEDV的有效疫苗,因此,治疗与诊断对防控PED具有重大意义。
抗体能够中和病毒,起到一定的治疗作用,但同时也具有一定的风险,比如产生ADE现象[22]。纳米抗体优越的生物学特性能够克服常规抗体的不足,很适合疫病的治疗[23-24]。Bao等[25]通过利用截短的PEDV S蛋白免疫骆驼后筛选出1株S蛋白特异性的纳米抗体S7,但S7对PEDV没有中和效果。Yang等[15]筛选出靶向PEDV M蛋白的4株特异性的单域抗体,但对PEDV同样没有中和活性。虽然没有中和活性,但这些纳米抗体均可被用于PEDV诊断。
纳米抗体被广泛用于病原诊断中[26-29],比如新城疫病毒(Newcastle disease virus, NDV)[17,27]、猪细小病毒(porcine parvovirus, PPV)[28]、猪流感病毒(swine influenza virus, SIV)[29]和寨卡病毒(Zika virus)[26]。纳米抗体也被用于PEDV的诊断中,我们课题组已经建立了基于PEDV N蛋白纳米抗体的快速检测PEDV的方法[30]。本研究表达截短的PEDV S1(22—505 aa)蛋白,该区域不含有中和表位,但能够影响病毒的复制[8]。利用截短的S1蛋白免疫骆驼后构建了多样性良好的VHH噬菌体展示文库。经鉴定,该文库库容量为2.1×107,随机挑选96个单菌落,阳性率达到85%, 对其中的阳性单克隆进行测序,验证了该文库具有良好的多样性。进一步对文库进行了救援和3轮特异性淘选富集,并最终筛选出6株氨基酸序列不相同的纳米抗体,6株纳米抗体与猪圆环病毒Cap蛋白、猪伪狂犬病病毒gE蛋白均不存在交叉反应。随后,验证结合力最高的纳米抗体Nb3与PEDV具有良好的结合活性。
4 结 论
成功表达并纯化PEDV S1蛋白,免疫双峰驼后,构建多样性良好的噬菌体展示文库,并利用噬菌体展示技术成功筛选出6株特异性结合PEDV S1蛋白的纳米抗体。所筛选的特异性纳米抗体为PEDV的诊断和纳米抗体药物的研发提供了参考。
