GC-MS法测定纺织品中的氯乙酸
2021-09-27吴秀芳
文/吴秀芳
1 引言
氯乙酸是一种用途广泛的物质,农药工业上用作制取乐果、萘乙酸、硫氰醋酸、异茨酯、除莠剂等的中间体;染料工业中用于生产靛蓝和萘氨基乙醚类染料;在制药工业上用于制取合成咖啡碱、肾上腺素、氨基醋酸、维生素B6、金霉素等的中间体;还可用于制造羟甲基纤维素。同时氯乙酸也是一种毒性较大的物质,氯乙酸可以通过吸入、食用、经过皮肤吸收的途径侵入人体。氯乙酸酸雾可致眼部刺激症状和角膜灼伤,严重的话会导致失明。即使是低浓度该品酸雾经常接触,也会发生头痛、头晕现象。
纺织品中氯乙酸的来源主要有两种,一种是作为1,2-二氯乙烷的降解物而存在,另一种是作为染料中间体的残留而存在。目前国内纺织品中涉及氯乙酸测定的标准只有GB/T 37633—2019。GB/T 37633—2019以甲醇为萃取剂萃取1,2-二氯乙烷、氯乙醇和氯乙酸。1,2-二氯乙烷和氯乙醇可以直接进行测定,但是由于氯乙酸有氯原子和羧基两个官能团,极性较强,沸点较高,挥发性较差,若直接测定会造成色谱峰拖尾,且响应值不稳定,还有可能会腐蚀色谱柱和检测器,所以将氯乙酸进行酯化[1],然后用甲基叔丁基醚作为提取剂进行提取,水洗,调pH值。但由于甲基叔丁基醚与甲醇互溶,且酯在酸性条件下不稳定,有造成酯类化合物响应值较低和不稳定的风险。本文采用正己烷作为氯乙酸甲酯的提取剂,提高了酯化效率,反应后先调节pH值,再进行水洗干燥,选用合适的色谱柱,优化色谱条件,从而实现对纺织品中氯乙酸高准确度、高精密度、高灵敏度的测定。
2 试验部分
2.1 主要仪器及试剂
主要仪器:7890A/5975C安捷伦气相色谱质谱联用仪;HZS-HA恒温水浴振荡器;KQ5200B型超声波发生器(工作频率40kHz);电子天平;20mL具塞可密封玻璃瓶。
主要试剂:氯乙酸标准品(德国 Dr Ehrenstorfer 公司);色谱级甲醇、色谱级甲基叔丁基醚、色谱级正己烷。
2.2 标准工作溶液的配制
用甲醇配制浓度为500mg/L的氯乙酸标液,然后把标液配制成相应浓度的标线。
2.3 GC-MS条件
毛细管色谱柱:DB-624(30m×0.32mm×1.8μm);进样口温度:250℃;传输线温度:250℃;电离方式:EI;离子源温度:230℃;载气:高纯氦气(≥99.999%);载气流速:2mL/min;进样量:1μL;进样方式:不分流进样。
扫描方式:选择离子监测。
2.4 试验步骤
样品剪碎混匀后,称取1.000g样品于20mL具塞玻璃瓶中,加入10mL色谱级甲醇,常温超声提取30min。
准确移取上述萃取液5mL于具塞离心管中,逐滴缓慢加入0.9mL浓硫酸(≥92%),加入5mL正己烷,在50℃的恒温水浴中静置酯化60min,然后冰水浴冷却。加入2mL饱和碳酸钠溶液,漩涡混匀离心,弃去下层的水相,再加入2mL硫酸钠溶液(20g/L),漩涡混匀离心弃去下层的水相,加入2g无水硫酸钠吸水干燥,经有机滤膜过滤后,用GC-MS分析氯乙酸甲酯。
3 结果与讨论
3.1 仪器分析条件的优化
3.1.1 色谱柱的选择
试验采用不同极性的DB-17、DB-200和DB-624毛细管柱对1,2-二氯乙烷及其降解物进行分析。结果显示目标化合物在DB-17和DB-200柱流失严重,杂质干扰较多,在DB-624柱上保留时间合适,几乎没有杂质干扰,分离效果好。因此,试验采用DB-624(30m×0.32mm×1.8μm)作为色谱柱。
3.1.2 色谱条件的优化
采用DB-624毛细管色谱柱,以初始温度、升温速率、载气流速为主要影响因素,经过一系列的优化,综合考虑出峰时间、分离度和峰型,最终选择如下的色谱条件:
在上述条件下氯乙酸甲酯的色谱图见图1,质谱图见图2。

图1 氯乙酸的GC-MS图

图2 氯乙酸甲酯质谱图
3.2 氯乙酸甲酯提取剂的选择
选择甲基叔丁基醚、乙醚、正己烷、乙酸乙酯对氯乙酸甲酯进行提取,结果表明甲基叔丁基醚与甲醇互溶,漩涡混匀离心没有出现分层现象,在加入了6mL的饱和Na2CO3溶液后才有分层出现,Na2CO3用量8mL~10mL之间提取效果最好,但有机相仅有约3mL,且水相里甲基叔丁基醚的味道浓烈。舍去水相,再用20g/L的Na2SO4溶液洗涤有机相,获得的有机相更少。乙醚和乙酸乙酯对氯乙酸甲酯的提取结果和甲基叔丁基醚类似。用正己烷提取氯乙酸甲酯时,正己烷加入后体系分层明显,有机相体积约5mL,加入2mL饱和Na2CO3后有机相和水相分层明显,舍去水相,再用20g/L的Na2SO4溶液洗涤有机相,分层明显,得到有机相体积约5mL。其原因可能是乙酸乙酯、乙醚、甲基叔丁基醚都与甲醇有一定的互溶性,而且甲醇与水互溶,导致提取效果不如正己烷好。
3.3 水洗和调节pH值顺序的优化
标准中氯乙酸甲酯用正己烷提取后先用20g/L的Na2SO4溶液洗涤有机相,然后再用Na2CO3溶液调节体系的pH值,本文先用Na2CO3溶液调节体系的pH值,然后再用Na2SO4洗。进行3次平行试验后,对比两种顺序下氯乙酸的回收率,结果如图3所示。
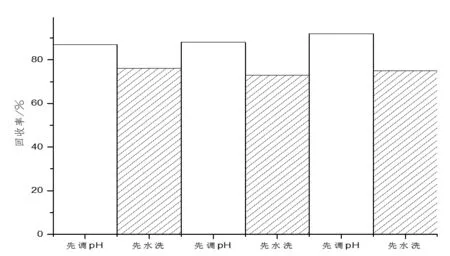
图3 氯乙酸甲酯不同后处理顺序回收率对比图
从图3中可以看出,先调节pH值的氯乙酸甲酯的回收率比先水洗的要高。分析可能是因为酯在酸性条件下不稳定。
3.4 线性方程与检出限
以色谱级甲醇为溶剂,称取适量的氯乙酸配制成质量浓度500mg/L的混合标准工作液,用甲醇逐级稀释配制为0.05mg/L~20mg/L的一系列标准工作液。
在优化好的色谱条件下,移取一系列标准曲线浓度的氯乙酸于离心管中,按照优化后的方法进行酯化,后处理。以氯乙酸甲酯的质量浓度(mg/L)为横坐标,特征离子吸收峰峰面积为纵坐标绘制标准曲线,得到氯乙酸甲酯的回归方程,结果如表1。
由表1可知,氯乙酸甲酯在0.05mg/L~20mg/L范围内,线性相关系数为0.9990,在较宽的浓度范围内有很好的线性相关系数。本方法的仪器检出限为0.05mg/kg。由表1可知,本试验有很低的方法检出限,表明本方法对氯乙酸的测定有较高的灵敏度。

表1 氯乙酸甲酯的线性方程和方法检出限
3.5 加标回收率和精密度
本试验以纯棉贴衬为空白样品进行加标回收,添加不同浓度的氯乙酸,每个浓度做6个平行样,用本文的前处理方法和仪器条件进行测试,氯乙酸回收率的数据和精密度如表2所示。

表2 氯乙酸甲酯回收率和精密度数据
由表2中的数据可知,氯乙酸甲酯回收率为86.7%~94.8%,相对标准偏差为5.0%~7.2%。可见优化后的方法对氯乙酸甲酯有很好的精密度和准确度。
3.6 实际样品分析
对不同基质的7个纺织品(棉织物、粘纤织物、聚酯织物、羊毛织物、锦纶织物、桑蚕丝织物、腈纶织物各3个)进行分析,结果均未检出氯乙酸。
4 结论
本文采用优化后的色谱方法,以正己烷为提取剂对纺织品中酯化后的氯乙酸进行提取和测定,优化后的方法提取效果、精密度和准确度都较好,适用于纺织品中氯乙酸的监测。
