绝对硬度的量化探究
——介绍一个计算化学实验
2021-09-26杨松汤义涵徐嘉伟刘偌曦薛冰张致慧
杨松,汤义涵,徐嘉伟,刘偌曦,薛冰,张致慧,*
1常州大学石油化工学院,江苏 常州 213164
2南京师范大学化学与材料科学学院,南京 210023
软硬酸碱(HSAB)理论是无机化学中的重要理论,可以预测物质的溶解度、化学反应的方向、判断配位化合物的稳定性。现行的无机化学教科书通常只给出软硬酸碱的分类依据,但并未定量地对其进行排序。目前,随着计算机技术的飞速发展,以及计算机技术的普及,计算化学逐步被非物理化学专业研究生以及高年级本科生所接受并使用。国内相关同行也开设了一些针对物理化学的计算化学本科生实验[1,2]。笔者在教学过程中,利用Gaussian 16软件,结合计算化学方法,设计实验研究化学硬度,帮助学生定量地理解这一概念。
1 实验目的
(1) 初步掌握对小分子进行单点能计算、几何构型优化和频率分析的基本操作;
(2) 掌握利用对称性加速计算的技巧;
(3) 掌握化学硬度η的计算公式。
2 实验原理
2.1 量子化学软件与基础知识
Gaussian是目前最常用的量子化学计算软件[3],最新版本是2016年的C.01版。其功能十分强大,可以完成几何优化、振动分析、势能面扫描、计算NMR等多种任务。本实验采用的是Windows系统下的软件,相比Linux系统而言,几乎不需要专业的计算机知识。该软件的输入文件以*.gjf结尾(gaussian job file),输出文件以*.out或*.log结尾。此外还可以通过%chk关键词产生*.chk文件,存储计算时产生的重要数据,例如能量、结构、分子轨道等,为后续的波函数分析提供依据。
量子化学计算的精度是由理论方法和基组共同决定的。密度泛函理论(Density Functional Theory,DFT)是目前主流的一类理论方法,其理论核心在于:体系基态的能量、波函数以及体系的各种性质,都是体系基态的电子密度分布的泛函。
对于常规分子体系,常用于几何优化和振动分析的泛函有B3LYP、M06-2X和PBE0等几种[4,5]。由于密度泛函具有一定的经验性(但不能因此将其认为是半经验的),不同的泛函在处理同一问题时得到的结果有时来去甚大,对于不同的体系需要根据经验选择不同的泛函。这样的经验可以从大量的测试数据中总结得出,但也不乏有一些问题是泛函处理存在劣势的。
基组是指一系列用于逼近真实原子轨道的数学函数,其本身可以不具有特定的化学意义。每种基组对其所适用的元素定义了计算时使用的基函数及基函数的组合方法。对于交换-相关泛函的一般性计算,常用的Ahlrichs系列基组大小如下:def2-TZVPP > def2-TZVP > def-TZVP > def2-SVP > def2-SV(P),如果是进行单点计算,在算力允许的情况下应当尽量选择较大基组。但如果是较耗时的几何优化或者振动分析,选择def-TZVP及以下档次的基组便足够。除此以外较为著名的还有Pople系列基组、pcseg系列基组以及本文使用的Dunning’s相关一致性基组,读者可自行查阅相关资料进一步了解。
本实验在M06-2X/def2-TZVP和CCSD(T)/aug-cc-pVQZ计算级别下进行。计算级别是指特定理论方法和基组的结合,可表示为“理论方法/基组”的形式。由Truhlar等人提出的M06-2X理论方法属于交换-相关泛函[6],从原理上来说,只要交换-相关泛函是精确的,那么结果也是精确的。CCSD(T)理论方法属于耦合簇方法(CC)[7],它通过微扰方式考虑更高一级激发算符,虽然是CCSDT (CC Singles,Doubles and Triples)的近似形式[8],但相对于CCSD (CC Singles and Doubles)而言仍有很大改进,很适合高精度计算,不过计算量巨大。def2-TZVP基组是Ahlrichs的def2系列基组的一种[9],属于3-ζ基组。其中def表示default,VP表示Valance Polazried。aug-cc-pVQZ是Dunning’s相关一致性基组[10,11],属于4-ζ基组,其中的aug表示对每个角动量都增加弥散函数。该基组应当结合CCSD(T)这类的后HF方法使用,尤其适合负离子体系的计算。
2.2 对称性在计算化学中的应用
以水分子为例,假设其并非C2υ点群,则需要两个O―H键长,一个H―O―H键角描述其结构。如果在建模时就限制其为C2υ点群,则仅需一个O―H键长,一个H―O―H键角描述,可有效减少计算量。对于苯(C6H6)这类略大的体系,如果采用高精度计算级别,耗时巨大。但考虑到它结构高度对称,属于D6h点群,开启对称性可以很大程度减少描述其结构所需参量,大大降低计算耗时,即便不使用服务器也可以提高计算能力,起到四两拨千斤的效果。
2.3 软硬酸碱理论
Pearson等人于1963年提出软硬酸碱理论[12]。在最初的定性分类中,“硬”指的是具有较小半径、电荷密度较高的物种,“软”指的是较大半径、电荷密度较低的物种。Pearson根据经验总结出“硬亲硬、软亲软”的软硬酸碱作用原理。
1968年,Klopman根据前线轨道中的微扰理论[13],对HSAB理论进行了初步的定量解释。他将软硬酸结合反应分成电荷控制和轨道控制两种。认为硬酸和硬碱结合受到电荷控制,软酸和软碱结合受到前线轨道能量控制。
1983年,Parr和Pearson进一步完善了前人的工作,提出了“绝对硬度”的概念[14],其定义式如下:

其中I代表电离能,A代表电子亲合能,根据此式可定量地比较物种的“硬度”。
3 实验所需软件和设备
Gaussian 16软件(C.01版)、GaussView 6.0软件[15]以及台式电脑(处理器:i7-9750H;核心数:12;运行内存:16GB)。
4 实验过程
本实验分为以下几步:
(1) 在M06-2X/def2-TZVP级别下优化中性分子(N电子)结构,并进行振动频率分析;
(2) 在M06-2X/def2-TZVP级别下分别对N电子分子、N± 1电子分子进行单点能计算(基于(1)所获取的结构),并记录耗时(注:虽然上一步计算会给出电子能量,但是为了让学生更直观地对比不同计算级别耗时的差异,增加了这一步单独进行单点能计算);
(3) 在CCSD(T)/aug-cc-pVQZ级别下分别对N电子分子、N± 1电子分子进行单点能计算(基于(1)所获取的结构),并记录耗时;
(4) 从单点能输出文件中读取电子能量,计算VIP和VEA,并根据公式(1)计算化学硬度η(课后完成,在实验报告中写出计算过程)。
要求学生计算H2O、CH3O−和CN−的化学硬度。
4.1 分子结构建模
打开Gaussview 6.0软件,依次点击“File”-“New Molecule Group”,绘制需要的结构(以水分子为例)。随后点击“Tools”-“Atom Groups”,开始进行对称性设定。点击“Enable Point Group Symmetry”启用对称性,在“Constrain to subgroup”中选择C2υ点群,最后勾选“Always track point group symmetry”,完成建模。点击“File”-“Save…”保存文件,并按要求更改文件名。
4.2 编写输入文件
在文件管理窗口选中刚刚保存的*.gjf文件,右击,选择以记事本方式打开。此时该输入文件应当如表1所示。
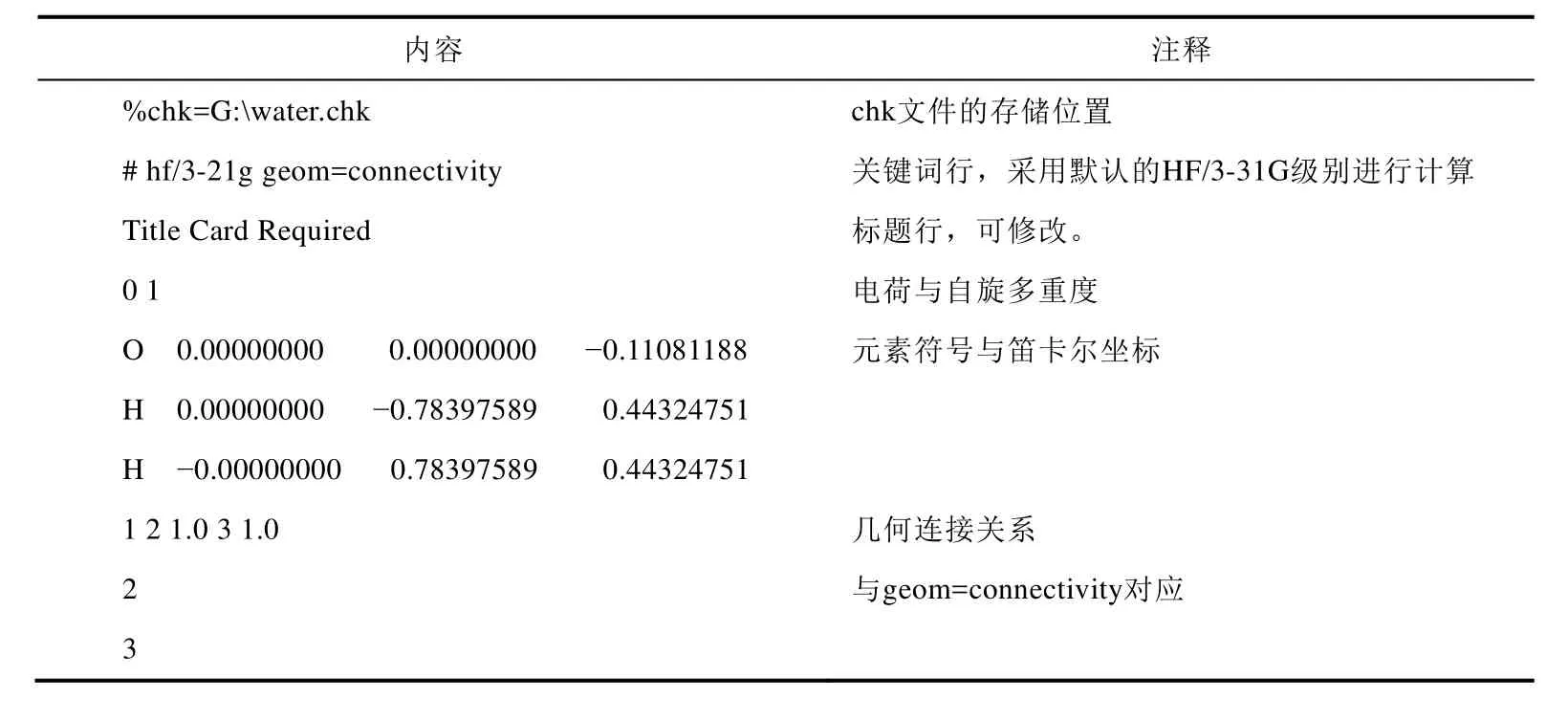
表1 输入文件内容及含义
实验第一步是在M06-2X/def2-TZVP级别下进行几何优化与振动频率分析,需要增加opt和freq关键词(分别对应optimization和frequency),并修改计算级别。同时可以删除几何连接关系,使得输入文件更加简洁。经过修改过的输入文件应当如表2所示。
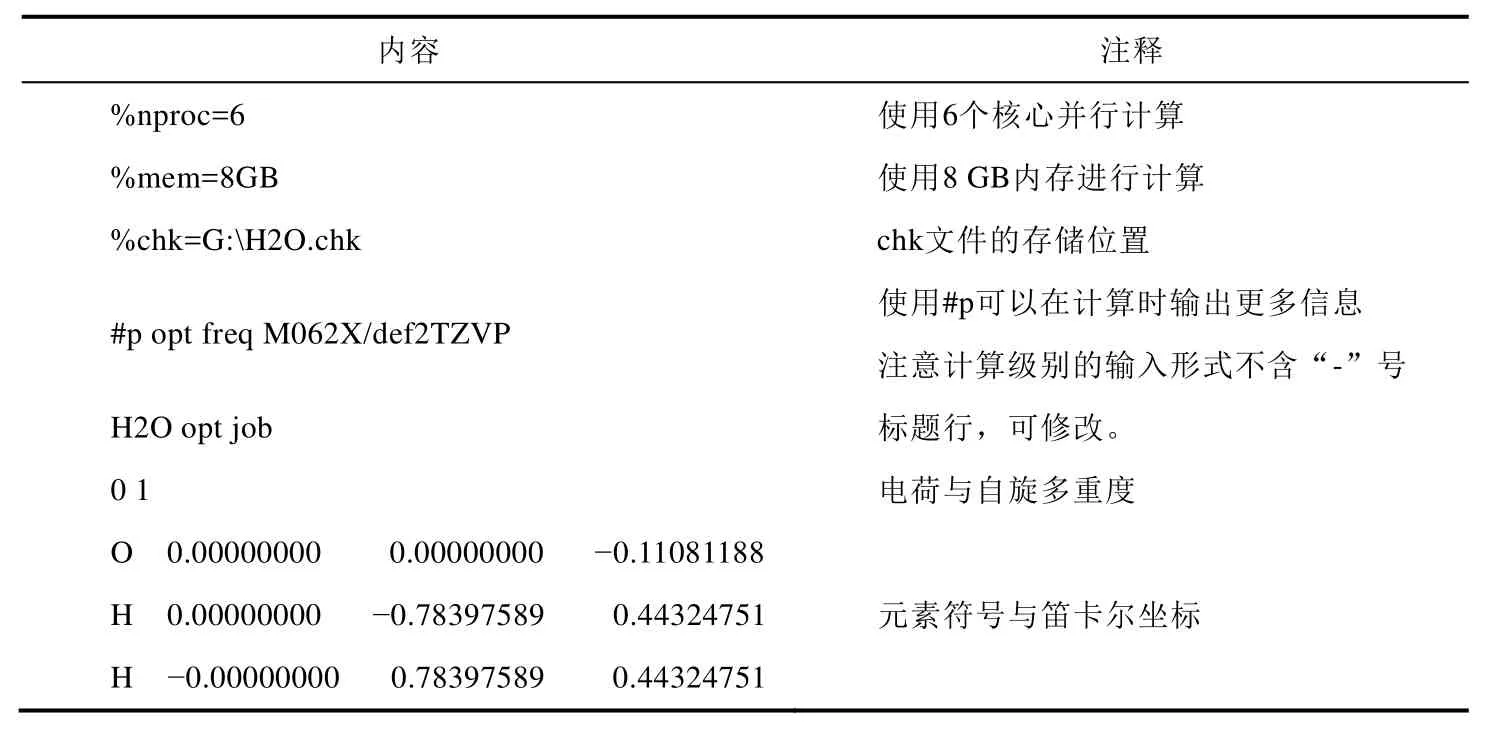
表2 经过修改过的输入文件内容及含义
一般而言,运行单一计算任务的时候,应当至少留出2 GB内存给系统,剩余内存都可以用来计算,分配给Gaussian的核心数一般等于计算机的物理核心数。在Gaussian的官方手册——《探寻化学的奥秘:电子结构方法》介绍了关于CPU使用量和研究问题大小的关系,感兴趣的同学可以自行阅读[16]。
保存修改过的输入文件。打开Gaussian 16软件,依次点击“File”-“New”-“File”-“Load”,选中刚才的输入文件,最后点击“Run”,选择输出文件的保存位置即可完成提交计算任务。待屏幕显示“Normal termination of Gaussian 16”时,表示计算正常结束。
使用Gaussview 6.0打开输出文件,右击,选择“Results”-“Vibrations”,可查看频率分析的结果。若无虚频,则已优化到了稳定结构,可以进行后续操作。将优化过后的水分子保存为三个新的输入文件,M06-2X/def2-TZVP级别下,依次进行H2O、H2O+和H2O−的单点能计算任务,该任务仅需指明计算级别即可,无需额外关键词。注意H2O+、H2O−是失去或得到一个电子的水分子,电荷为分别为+1和−1,自旋多重度为2。
完成上一步骤后,将优化过的水分子再保存为三个新的输入文件,在CCSD(T)/aug-cc-pVQZ级别下,依次进行H2O、H2O+和H2O–的单点能计算任务。注意计算级别的输入形式与“CCSD(T)/aug-ccpVQZ”完全一致。
4.3 读取计算结果
选中单点能计算输出结果的*.out文件,右击,选择以记事本方式打开,在输出文件的末尾有所需的电子能量信息。M06-2X/def2-TZVP级别下进行的计算,应当在“HF = ”处读取。CCSD(T)/augcc-pVQZ级别下进行的计算,应当在“CCSD(T) = ”处读取。
5 数据处理
整理本实验的单点计算步骤所获取数据至表3 (以H2O为例)。

表3 H2O的单点能计算结果汇总表
对CH3O−和CN−进行同样的操作,根据式(1),计算化学硬度并汇总至表4。

表4 化学硬度计算汇总表
6 思考与延展阅读
(1) 对O2进行计算时,O2的自旋多重度应该是多少?
答:根据分子轨道理论,O2的基态价电子组态为(2σg)2(2σu)2(3σg)2(1πu)4(1πg)2,有两个自旋方向相同的单电子(n= 2),故自旋多重度S= 3。
(2) 实验过程中的(1)、(2)步,或(1)、(3)步,能否使用热力学组合方法,诸如G4等方法一步完成?
答:不能,热力学组合方法是一套组合过程,包含几何优化、频率分析和高级别单点能计算等步骤。计算VIP或VEA时,假定分子结构是不变的,如果使用热力学组合方法,会自动优化结构,从而改变原始结构。但当计算绝热电离能(AIP)或绝热电子亲合能(AEA)时,要求分别对N,N± 1电子结构进行优化,则可以使用热力学组合方法,更加方便。
(3) Koopmans定理指出,VIP可由HOMO轨道能量近似代替,VEA可由LUMO轨道能量近似代替,也就是说进行几何结构优化,获取分子轨道能量后即可计算绝对硬度。请你读取有关数据,根据Koopmans定理计算,看看结果与实验值的误差。
(4) 从计算结果来看,高精度级别可以获得准确性较高的数据,但需要付出高昂的时间成本。这告诉我们,在今后进行实际研究时,需要根据算力和体系大小选择合适的计算级别,以较少的时间成本获取最经济的数据。
7 结语
本实验侧重于使用计算化学方法解决实际问题,而不过分追求学生对其中量子化学原理的深入理解。因为受众是高年级本科生和非计算化学专业的研究生,并不具备相关专业知识。但只要学生具备无机化学和结构化学的基本知识,以及一定的计算机操作技能,便可利用计算化学方法辅助研究。这需要教师具备一定的计算化学知识,对于计算级别的选取,计算过程中出现的错误,能够提供指导并及时解决。
笔者认为,开设这样的探究型实验有以下好处:
(1) 激发学生进行验证课本知识以及进行科学研究的兴趣。例如,氧气的第一电离能测定实验,大多数学生是没有机会涉及的,但可以通过高精度计算获得和实验值相差很小的数据。学生在探究过程中能够获得满足感,体会到了科学研究的价值。再比如,无机化学教材中只简单地对软硬酸碱进行定性的分类,但没有对其进行定量的排序。学生通过本实验,可以定量地给出化学硬度的大小。
(2) 培养学科交叉人才。虽然本实验研究的是无机化学中几种常见配体的硬度,但是对于有机物来说计算的基本流程也是一样的。笔者计划在后续的课程中加入局部化学硬度的计算实验,可以用来判断多齿配体中不同配位原子进行配位的倾向,也可以为有机反应中的选择性提供指导。
目前,本实验已在笔者近年指导的江苏省大学生创新项目中开展,并在进一步完善丰富实验内容,计划向学校提交完整的实验课程方案,开设“计算化学在无机化学的应用”选修课程。同时笔者也已在所讲授的高等结构化学课程中通过Gaussview软件展示分子轨道形状,演示化学键的成键过程,帮助学生更直观地理解。本课题组还与美国乔治亚理工学院、中科院上海药物研究所、中科院高能物理研究所等高校或科研院所开展有效合作,培养了一批具备一定计算化学能力的高年级本科生和研究生。
